Publications: 2018
2018
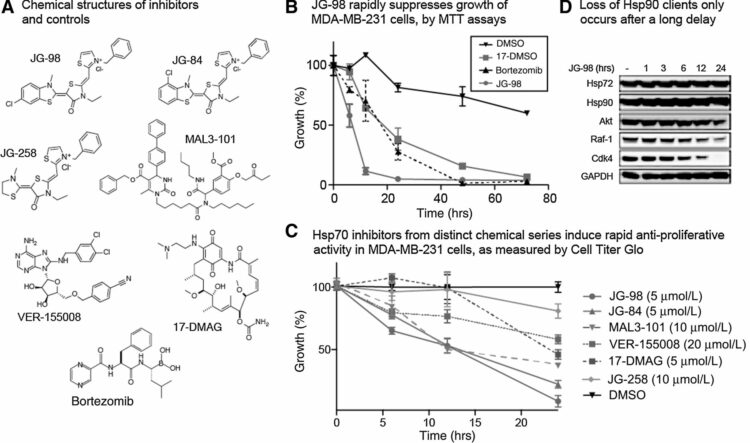
Sun, Jian; Paduch, Marcin; Kim, Sang-Ah; Kramer, Ryan M; Barrios, Adam F; Lu, Vincent; Luke, Judy; Usatyuk, Svitlana; Kossiakoff, Anthony A; Tan, Song
Structural basis for activation of SAGA histone acetyltransferase Gcn5 by partner subunit Ada2 Journal Article
In: Proc Natl Acad Sci U S A, vol. 115, no. 40, pp. 10010–10015, 2018, ISSN: 1091-6490.
@article{pmid30224453,
title = {Structural basis for activation of SAGA histone acetyltransferase Gcn5 by partner subunit Ada2},
author = {Jian Sun and Marcin Paduch and Sang-Ah Kim and Ryan M Kramer and Adam F Barrios and Vincent Lu and Judy Luke and Svitlana Usatyuk and Anthony A Kossiakoff and Song Tan},
doi = {10.1073/pnas.1805343115},
issn = {1091-6490},
year = {2018},
date = {2018-10-01},
urldate = {2018-10-01},
journal = {Proc Natl Acad Sci U S A},
volume = {115},
number = {40},
pages = {10010--10015},
abstract = {The Gcn5 histone acetyltransferase (HAT) subunit of the SAGA transcriptional coactivator complex catalyzes acetylation of histone H3 and H2B N-terminal tails, posttranslational modifications associated with gene activation. Binding of the SAGA subunit partner Ada2 to Gcn5 activates Gcn5's intrinsically weak HAT activity on histone proteins, but the mechanism for this activation by the Ada2 SANT domain has remained elusive. We have employed Fab antibody fragments as crystallization chaperones to determine crystal structures of a yeast Ada2/Gcn5 complex. Our structural and biochemical results indicate that the Ada2 SANT domain does not activate Gcn5's activity by directly affecting histone peptide binding as previously proposed. Instead, the Ada2 SANT domain enhances Gcn5 binding of the enzymatic cosubstrate acetyl-CoA. This finding suggests a mechanism for regulating chromatin modification enzyme activity: controlling binding of the modification cosubstrate instead of the histone substrate.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
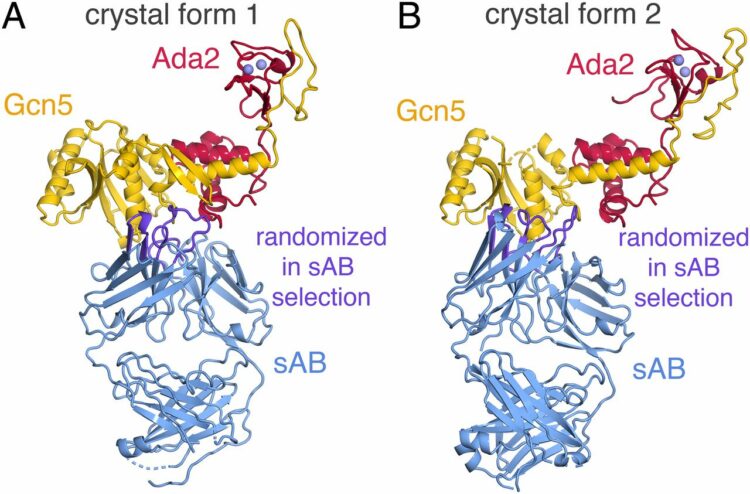
Kintzer, Alexander F; Green, Evan M; Dominik, Pawel K; Bridges, Michael; Armache, Jean-Paul; Deneka, Dawid; Kim, Sangwoo S; Hubbell, Wayne; Kossiakoff, Anthony A; Cheng, Yifan; Stroud, Robert M
Structural basis for activation of voltage sensor domains in an ion channel TPC1 Journal Article
In: Proc Natl Acad Sci U S A, vol. 115, no. 39, pp. E9095–E9104, 2018, ISSN: 1091-6490.
@article{pmid30190435,
title = {Structural basis for activation of voltage sensor domains in an ion channel TPC1},
author = {Alexander F Kintzer and Evan M Green and Pawel K Dominik and Michael Bridges and Jean-Paul Armache and Dawid Deneka and Sangwoo S Kim and Wayne Hubbell and Anthony A Kossiakoff and Yifan Cheng and Robert M Stroud},
doi = {10.1073/pnas.1805651115},
issn = {1091-6490},
year = {2018},
date = {2018-09-01},
urldate = {2018-09-01},
journal = {Proc Natl Acad Sci U S A},
volume = {115},
number = {39},
pages = {E9095--E9104},
abstract = {Voltage-sensing domains (VSDs) couple changes in transmembrane electrical potential to conformational changes that regulate ion conductance through a central channel. Positively charged amino acids inside each sensor cooperatively respond to changes in voltage. Our previous structure of a TPC1 channel captured an example of a resting-state VSD in an intact ion channel. To generate an activated-state VSD in the same channel we removed the luminal inhibitory Ca-binding site (Ca), which shifts voltage-dependent opening to more negative voltage and activation at 0 mV. Cryo-EM reveals two coexisting structures of the VSD, an intermediate state 1 that partially closes access to the cytoplasmic side but remains occluded on the luminal side and an intermediate activated state 2 in which the cytoplasmic solvent access to the gating charges closes, while luminal access partially opens. Activation can be thought of as moving a hydrophobic insulating region of the VSD from the external side to an alternate grouping on the internal side. This effectively moves the gating charges from the inside potential to that of the outside. Activation also requires binding of Ca to a cytoplasmic site (Ca). An X-ray structure with Ca removed and a near-atomic resolution cryo-EM structure with Ca removed define how dramatic conformational changes in the cytoplasmic domains may communicate with the VSD during activation. Together four structures provide a basis for understanding the voltage-dependent transition from resting to activated state, the tuning of VSD by thermodynamic stability, and this channel's requirement of cytoplasmic Ca ions for activation.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
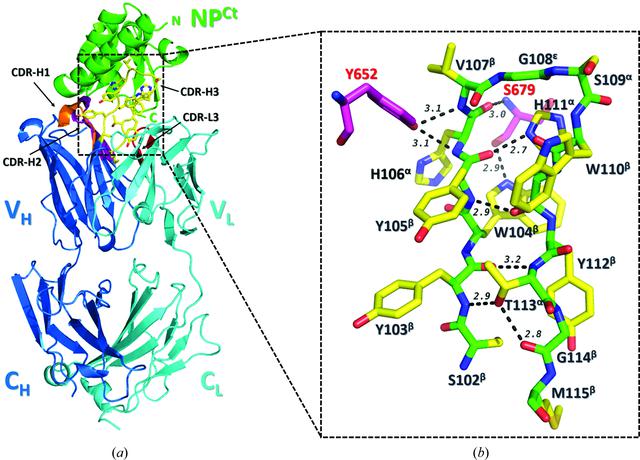
Radwańska, Malwina J; Jaskółowski, Mateusz; Davydova, Elena; Derewenda, Urszula; Miyake, Tsuyoshi; Engel, Daniel A; Kossiakoff, Anthony A; Derewenda, Zygmunt S
In: Acta Crystallogr D Struct Biol, vol. 74, no. Pt 7, pp. 681–689, 2018, ISSN: 2059-7983.
@article{pmid29968677,
title = {The structure of the C-terminal domain of the nucleoprotein from the Bundibugyo strain of the Ebola virus in complex with a pan-specific synthetic Fab},
author = {Malwina J Radwańska and Mateusz Jaskółowski and Elena Davydova and Urszula Derewenda and Tsuyoshi Miyake and Daniel A Engel and Anthony A Kossiakoff and Zygmunt S Derewenda},
doi = {10.1107/S2059798318007878},
issn = {2059-7983},
year = {2018},
date = {2018-07-01},
urldate = {2018-07-01},
journal = {Acta Crystallogr D Struct Biol},
volume = {74},
number = {Pt 7},
pages = {681--689},
abstract = {The vast majority of platforms for the detection of viral or bacterial antigens rely on immunoassays, typically ELISA or sandwich ELISA, that are contingent on the availability of suitable monoclonal antibodies (mAbs). This is a major bottleneck, since the generation and production of mAbs is time-consuming and expensive. Synthetic antibody fragments (sFabs) generated by phage-display selection offer an alternative with many advantages over Fabs obtained from natural antibodies using hybridoma technology. Unlike mAbs, sFabs are generated using phage display, allowing selection for binding to specific strains or for pan-specificity, for identification of structural epitopes or unique protein conformations and even for complexes. Further, they can easily be produced in Escherichia coli in large quantities and engineered for purposes of detection technologies and other applications. Here, the use of phage-display selection to generate a pan-specific Fab (MJ20), based on a Herceptin Fab scaffold, with the ability to bind selectively and with high affinity to the C-terminal domains of the nucleoproteins (NPs) from all five known strains of the Ebola virus is reported. The high-resolution crystal structure of the complex of MJ20 with the antigen from the Bundibugyo strain of the Ebola virus reveals the basis for pan-specificity and illustrates how the phage-display technology can be used to manufacture suitable Fabs for use in diagnostic or therapeutic applications.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Kang, Yanyong; Kuybeda, Oleg; de Waal, Parker W; Mukherjee, Somnath; Eps, Ned Van; Dutka, Przemyslaw; Zhou, X Edward; Bartesaghi, Alberto; Erramilli, Satchal; Morizumi, Takefumi; Gu, Xin; Yin, Yanting; Liu, Ping; Jiang, Yi; Meng, Xing; Zhao, Gongpu; Melcher, Karsten; Ernst, Oliver P; Kossiakoff, Anthony A; Subramaniam, Sriram; Xu, H Eric
Cryo-EM structure of human rhodopsin bound to an inhibitory G protein Journal Article
In: Nature, vol. 558, no. 7711, pp. 553–558, 2018, ISSN: 1476-4687.
@article{pmid29899450,
title = {Cryo-EM structure of human rhodopsin bound to an inhibitory G protein},
author = {Yanyong Kang and Oleg Kuybeda and Parker W de Waal and Somnath Mukherjee and Ned Van Eps and Przemyslaw Dutka and X Edward Zhou and Alberto Bartesaghi and Satchal Erramilli and Takefumi Morizumi and Xin Gu and Yanting Yin and Ping Liu and Yi Jiang and Xing Meng and Gongpu Zhao and Karsten Melcher and Oliver P Ernst and Anthony A Kossiakoff and Sriram Subramaniam and H Eric Xu},
doi = {10.1038/s41586-018-0215-y},
issn = {1476-4687},
year = {2018},
date = {2018-06-01},
urldate = {2018-06-01},
journal = {Nature},
volume = {558},
number = {7711},
pages = {553--558},
abstract = {G-protein-coupled receptors comprise the largest family of mammalian transmembrane receptors. They mediate numerous cellular pathways by coupling with downstream signalling transducers, including the hetrotrimeric G proteins G (stimulatory) and G (inhibitory) and several arrestin proteins. The structural mechanisms that define how G-protein-coupled receptors selectively couple to a specific type of G protein or arrestin remain unknown. Here, using cryo-electron microscopy, we show that the major interactions between activated rhodopsin and G are mediated by the C-terminal helix of the G α-subunit, which is wedged into the cytoplasmic cavity of the transmembrane helix bundle and directly contacts the amino terminus of helix 8 of rhodopsin. Structural comparisons of inactive, G-bound and arrestin-bound forms of rhodopsin with inactive and G-bound forms of the β-adrenergic receptor provide a foundation to understand the unique structural signatures that are associated with the recognition of G, G and arrestin by activated G-protein-coupled receptors.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
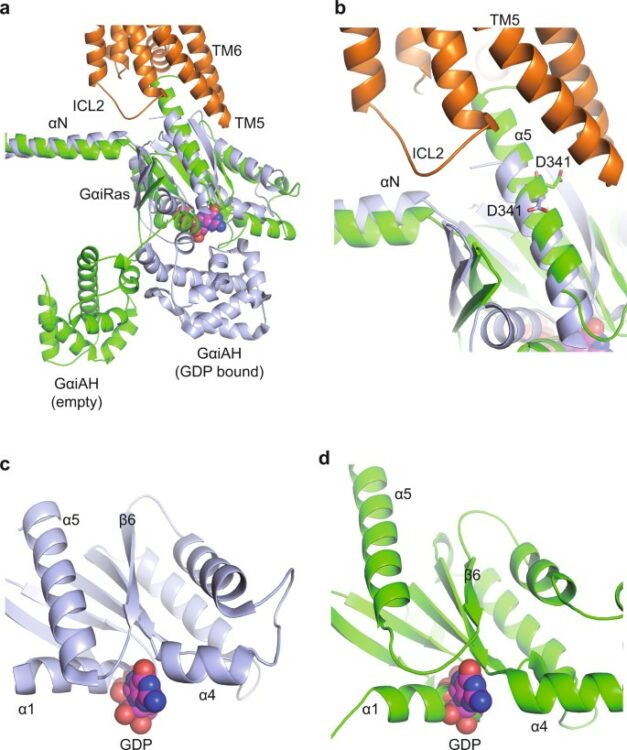
Keedy, Daniel A; Hill, Zachary B; Biel, Justin T; Kang, Emily; Rettenmaier, T Justin; Brandão-Neto, José; Pearce, Nicholas M; von Delft, Frank; Wells, James A; Fraser, James S
An expanded allosteric network in PTP1B by multitemperature crystallography, fragment screening, and covalent tethering Journal Article
In: Elife, vol. 7, 2018, ISSN: 2050-084X.
@article{pmid29877794,
title = {An expanded allosteric network in PTP1B by multitemperature crystallography, fragment screening, and covalent tethering},
author = {Daniel A Keedy and Zachary B Hill and Justin T Biel and Emily Kang and T Justin Rettenmaier and José Brandão-Neto and Nicholas M Pearce and Frank von Delft and James A Wells and James S Fraser},
doi = {10.7554/eLife.36307},
issn = {2050-084X},
year = {2018},
date = {2018-06-01},
urldate = {2018-06-01},
journal = {Elife},
volume = {7},
abstract = {Allostery is an inherent feature of proteins, but it remains challenging to reveal the mechanisms by which allosteric signals propagate. A clearer understanding of this intrinsic circuitry would afford new opportunities to modulate protein function. Here, we have identified allosteric sites in protein tyrosine phosphatase 1B (PTP1B) by combining multiple-temperature X-ray crystallography experiments and structure determination from hundreds of individual small-molecule fragment soaks. New modeling approaches reveal 'hidden' low-occupancy conformational states for protein and ligands. Our results converge on allosteric sites that are conformationally coupled to the active-site WPD loop and are hotspots for fragment binding. Targeting one of these sites with covalently tethered molecules or mutations allosterically inhibits enzyme activity. Overall, this work demonstrates how the ensemble nature of macromolecular structure, revealed here by multitemperature crystallography, can elucidate allosteric mechanisms and open new doors for long-range control of protein function.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Huang, Ganggang; Zhong, Zhenwei; Miersch, Shane; Sidhu, Sachdev S; Hou, Shin-Chen; Wu, Donghui
Construction of Synthetic Phage Displayed Fab Library with Tailored Diversity Journal Article
In: J Vis Exp, no. 135, 2018, ISSN: 1940-087X.
@article{pmid29782009,
title = {Construction of Synthetic Phage Displayed Fab Library with Tailored Diversity},
author = {Ganggang Huang and Zhenwei Zhong and Shane Miersch and Sachdev S Sidhu and Shin-Chen Hou and Donghui Wu},
doi = {10.3791/57357},
issn = {1940-087X},
year = {2018},
date = {2018-05-01},
urldate = {2018-05-01},
journal = {J Vis Exp},
number = {135},
abstract = {Demand for monoclonal antibodies (mAbs) in basic research and medicine is increasing yearly. Hybridoma technology has been the dominant method for mAb development since its first report in 1975. As an alternative technology, phage display methods for mAb development are increasingly attractive since Humira, the first phage-derived antibody and one of the best-selling mAbs, was approved for clinical treatment of rheumatoid arthritis in 2002. As a non-animal based mAb development technology, phage display bypasses antigen immunogenicity, humanization, and animal maintenance that are required from traditional hybridoma technology based antibody development. In this protocol, we describe a method for construction of synthetic phage-displayed Fab libraries with diversities of 10-10 obtainable with a single electroporation. This protocol consists of: 1) high-efficiency electro-competent cell preparation; 2) extraction of uracil-containing single-stranded DNA (dU-ssDNA); 3) Kunkel's method based oligonucleotide-directed mutagenesis; 4) electroporation and calculation of library size; 5) protein A/L-based enzyme-linked immunosorbent assay (ELISA) for folding and functional diversity evaluation; and 6) DNA sequence analysis of diversity.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
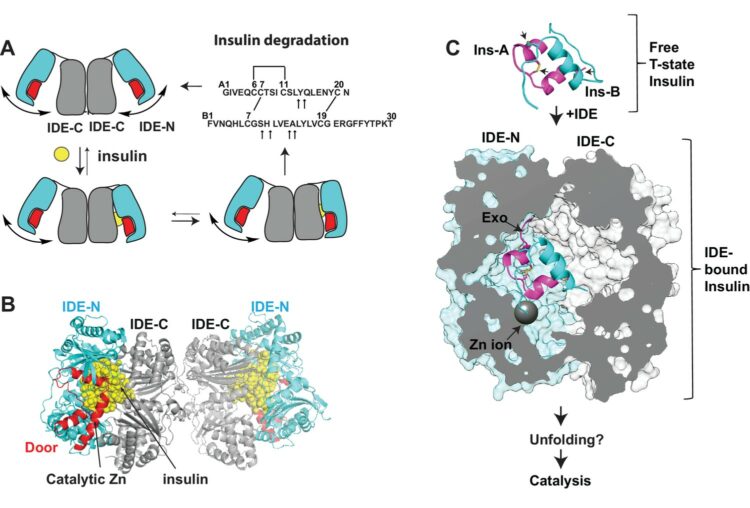
Zhang, Zhening; Liang, Wenguang G; Bailey, Lucas J; Tan, Yong Zi; Wei, Hui; Wang, Andrew; Farcasanu, Mara; Woods, Virgil A; McCord, Lauren A; Lee, David; Shang, Weifeng; Deprez-Poulain, Rebecca; Deprez, Benoit; Liu, David R; Koide, Akiko; Koide, Shohei; Kossiakoff, Anthony A; Li, Sheng; Carragher, Bridget; Potter, Clinton S; Tang, Wei-Jen
Ensemble cryoEM elucidates the mechanism of insulin capture and degradation by human insulin degrading enzyme Journal Article
In: Elife, vol. 7, 2018, ISSN: 2050-084X.
@article{pmid29596046,
title = {Ensemble cryoEM elucidates the mechanism of insulin capture and degradation by human insulin degrading enzyme},
author = {Zhening Zhang and Wenguang G Liang and Lucas J Bailey and Yong Zi Tan and Hui Wei and Andrew Wang and Mara Farcasanu and Virgil A Woods and Lauren A McCord and David Lee and Weifeng Shang and Rebecca Deprez-Poulain and Benoit Deprez and David R Liu and Akiko Koide and Shohei Koide and Anthony A Kossiakoff and Sheng Li and Bridget Carragher and Clinton S Potter and Wei-Jen Tang},
doi = {10.7554/eLife.33572},
issn = {2050-084X},
year = {2018},
date = {2018-03-01},
urldate = {2018-03-01},
journal = {Elife},
volume = {7},
abstract = {Insulin degrading enzyme (IDE) plays key roles in degrading peptides vital in type two diabetes, Alzheimer's, inflammation, and other human diseases. However, the process through which IDE recognizes peptides that tend to form amyloid fibrils remained unsolved. We used cryoEM to understand both the apo- and insulin-bound dimeric IDE states, revealing that IDE displays a large opening between the homologous ~55 kDa N- and C-terminal halves to allow selective substrate capture based on size and charge complementarity. We also used cryoEM, X-ray crystallography, SAXS, and HDX-MS to elucidate the molecular basis of how amyloidogenic peptides stabilize the disordered IDE catalytic cleft, thereby inducing selective degradation by substrate-assisted catalysis. Furthermore, our insulin-bound IDE structures explain how IDE processively degrades insulin by stochastically cutting either chain without breaking disulfide bonds. Together, our studies provide a mechanism for how IDE selectively degrades amyloidogenic peptides and offers structural insights for developing IDE-based therapies.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Pollock, Samuel B; Hu, Amy; Mou, Yun; Martinko, Alexander J; Julien, Olivier; Hornsby, Michael; Ploder, Lynda; Adams, Jarrett J; Geng, Huimin; Müschen, Markus; Sidhu, Sachdev S; Moffat, Jason; Wells, James A
Highly multiplexed and quantitative cell-surface protein profiling using genetically barcoded antibodies Journal Article
In: Proc Natl Acad Sci U S A, vol. 115, no. 11, pp. 2836–2841, 2018, ISSN: 1091-6490.
@article{pmid29476010,
title = {Highly multiplexed and quantitative cell-surface protein profiling using genetically barcoded antibodies},
author = {Samuel B Pollock and Amy Hu and Yun Mou and Alexander J Martinko and Olivier Julien and Michael Hornsby and Lynda Ploder and Jarrett J Adams and Huimin Geng and Markus Müschen and Sachdev S Sidhu and Jason Moffat and James A Wells},
doi = {10.1073/pnas.1721899115},
issn = {1091-6490},
year = {2018},
date = {2018-03-01},
urldate = {2018-03-01},
journal = {Proc Natl Acad Sci U S A},
volume = {115},
number = {11},
pages = {2836--2841},
abstract = {Human cells express thousands of different surface proteins that can be used for cell classification, or to distinguish healthy and disease conditions. A method capable of profiling a substantial fraction of the surface proteome simultaneously and inexpensively would enable more accurate and complete classification of cell states. We present a highly multiplexed and quantitative surface proteomic method using genetically barcoded antibodies called phage-antibody next-generation sequencing (PhaNGS). Using 144 preselected antibodies displayed on filamentous phage (Fab-phage) against 44 receptor targets, we assess changes in B cell surface proteins after the development of drug resistance in a patient with acute lymphoblastic leukemia (ALL) and in adaptation to oncogene expression in a Myc-inducible Burkitt lymphoma model. We further show PhaNGS can be applied at the single-cell level. Our results reveal that a common set of proteins including FLT3, NCR3LG1, and ROR1 dominate the response to similar oncogenic perturbations in B cells. Linking high-affinity, selective, genetically encoded binders to NGS enables direct and highly multiplexed protein detection, comparable to RNA-sequencing for mRNA. PhaNGS has the potential to profile a substantial fraction of the surface proteome simultaneously and inexpensively to enable more accurate and complete classification of cell states.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
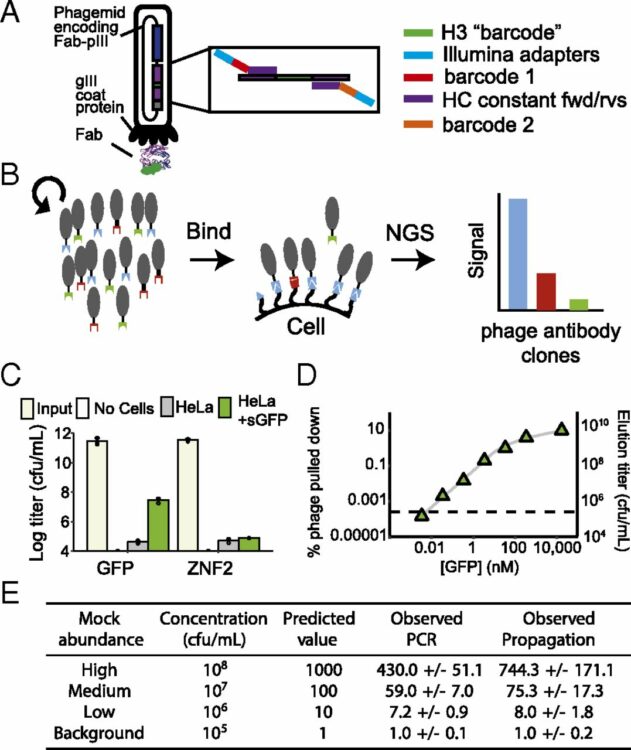
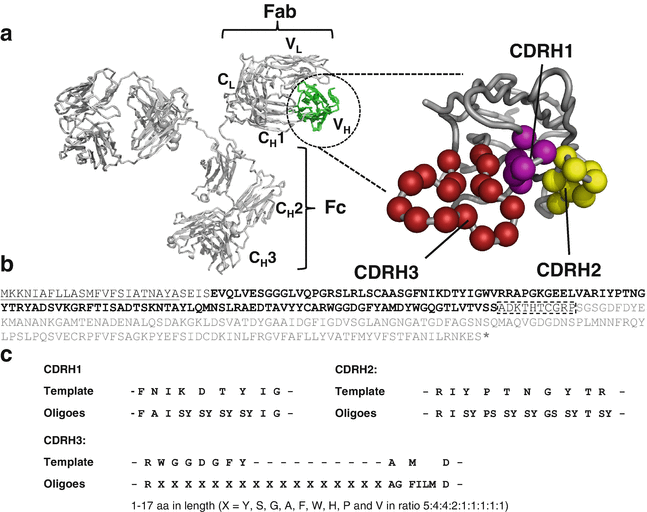
Bailey, Lucas J; Sheehy, Kimberly M; Dominik, Pawel K; Liang, Wenguang G; Rui, Huan; Clark, Michael; Jaskolowski, Mateusz; Kim, Yejoon; Deneka, Dawid; Tang, Wei-Jen; Kossiakoff, Anthony A
Locking the Elbow: Improved Antibody Fab Fragments as Chaperones for Structure Determination Journal Article
In: J Mol Biol, vol. 430, no. 3, pp. 337–347, 2018, ISSN: 1089-8638.
@article{pmid29273204,
title = {Locking the Elbow: Improved Antibody Fab Fragments as Chaperones for Structure Determination},
author = {Lucas J Bailey and Kimberly M Sheehy and Pawel K Dominik and Wenguang G Liang and Huan Rui and Michael Clark and Mateusz Jaskolowski and Yejoon Kim and Dawid Deneka and Wei-Jen Tang and Anthony A Kossiakoff},
doi = {10.1016/j.jmb.2017.12.012},
issn = {1089-8638},
year = {2018},
date = {2018-02-01},
urldate = {2018-02-01},
journal = {J Mol Biol},
volume = {430},
number = {3},
pages = {337--347},
abstract = {Antibody Fab fragments have been exploited with significant success to facilitate the structure determination of challenging macromolecules as crystallization chaperones and as molecular fiducial marks for single particle cryo-electron microscopy approaches. However, the inherent flexibility of the "elbow" regions, which link the constant and variable domains of the Fab, can introduce disorder and thus diminish their effectiveness. We have developed a phage display engineering strategy to generate synthetic Fab variants that significantly reduces elbow flexibility, while maintaining their high affinity and stability. This strategy was validated using previously recalcitrant Fab-antigen complexes where introduction of an engineered elbow region enhanced crystallization and diffraction resolution. Furthermore, incorporation of the mutations appears to be generally portable to other synthetic antibodies and may serve as a universal strategy to enhance the success rates of Fabs as structure determination chaperones.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
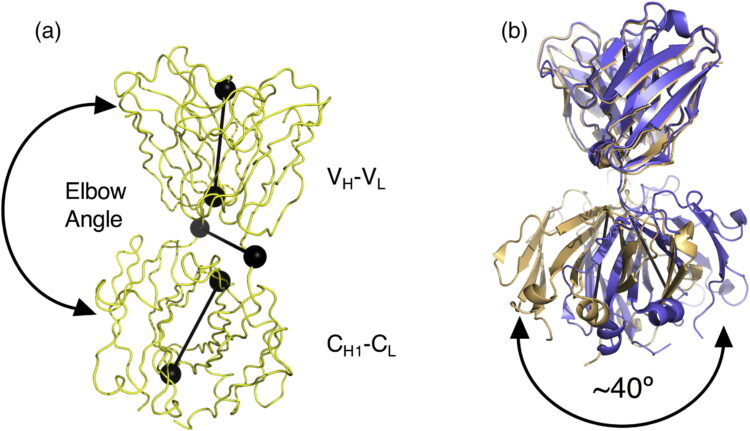
Mukherjee, Somnath; Griffin, Dionne H; Horn, James R; Rizk, Shahir S; Nocula-Lugowska, Malgorzata; Malmqvist, Magnus; Kim, Sangwoo S; Kossiakoff, Anthony A
Engineered synthetic antibodies as probes to quantify the energetic contributions of ligand binding to conformational changes in proteins Journal Article
In: J Biol Chem, vol. 293, no. 8, pp. 2815–2828, 2018, ISSN: 1083-351X.
@article{pmid29321208,
title = {Engineered synthetic antibodies as probes to quantify the energetic contributions of ligand binding to conformational changes in proteins},
author = {Somnath Mukherjee and Dionne H Griffin and James R Horn and Shahir S Rizk and Malgorzata Nocula-Lugowska and Magnus Malmqvist and Sangwoo S Kim and Anthony A Kossiakoff},
doi = {10.1074/jbc.RA117.000656},
issn = {1083-351X},
year = {2018},
date = {2018-02-01},
urldate = {2018-02-01},
journal = {J Biol Chem},
volume = {293},
number = {8},
pages = {2815--2828},
abstract = {Conformational changes in proteins due to ligand binding are ubiquitous in biological processes and are integral to many biological systems. However, it is often challenging to link ligand-induced conformational changes to a resulting biological function because it is difficult to distinguish between the energetic components associated with ligand binding and those due to structural rearrangements. Here, we used a unique approach exploiting conformation-specific and regio-specific synthetic antibodies (sABs) to probe the energetic contributions of ligand binding to conformation changes. Using maltose-binding protein (MBP) as a model system, customized phage-display selections were performed to generate sABs that stabilize MBP in different conformational states, modulating ligand-binding affinity in competitive, allosteric, or peristeric manners. We determined that the binding of a closed conformation-specific sAB (sAB-11M) to MBP in the absence of maltose is entropically driven, providing new insight into designing antibody-stabilized protein interactions. Crystal structures of sABs bound to MBP, together with biophysical data, delineate the basis of free energy differences between different conformational states and confirm the use of the sABs as energy probes for dissecting enthalpic and entropic contributions to conformational transitions. Our work provides a foundation for investigating the energetic contributions of distinct conformational dynamics to specific biological outputs. We anticipate that our approach also may be valuable for analyzing the energy landscapes of regulatory proteins controlling biological responses to environmental changes.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
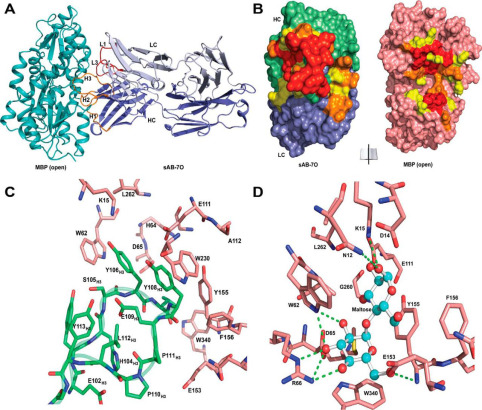
Hill, Zachary B; Martinko, Alexander J; Nguyen, Duy P; Wells, James A
Human antibody-based chemically induced dimerizers for cell therapeutic applications Journal Article
In: Nat Chem Biol, vol. 14, no. 2, pp. 112–117, 2018, ISSN: 1552-4469.
@article{pmid29200207,
title = {Human antibody-based chemically induced dimerizers for cell therapeutic applications},
author = {Zachary B Hill and Alexander J Martinko and Duy P Nguyen and James A Wells},
doi = {10.1038/nchembio.2529},
issn = {1552-4469},
year = {2018},
date = {2018-02-01},
urldate = {2018-02-01},
journal = {Nat Chem Biol},
volume = {14},
number = {2},
pages = {112--117},
abstract = {Chemically induced dimerizers (CIDs) have emerged as one of the most powerful tools for artificially regulating signaling pathways in cells; however, currently available CID systems lack the properties desired for use in regulating cellular therapies. Here, we report the development of human antibody-based chemically induced dimerizers (AbCIDs) from known small-molecule-protein complexes by selecting for synthetic antibodies that recognize the chemical epitope created by the bound small molecule. We demonstrate this concept by generating three antibodies that are highly selective for the BCL-xL-ABT-737 complex compared to BCL-xL alone. We show the potential of AbCIDs for application in regulating human cell therapies by using them to induce CRISPRa-mediated gene expression and to regulate CAR T-cell activation. We believe that the AbCIDs generated in this study will find application in regulating cell therapies and that the general method of AbCID development may lead to the creation of many new and orthogonal CIDs.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Nilvebrant, Johan; Sidhu, Sachdev S
Construction of Synthetic Antibody Phage-Display Libraries Journal Article
In: Methods Mol Biol, vol. 1701, pp. 45–60, 2018, ISSN: 1940-6029.
@article{pmid29116499,
title = {Construction of Synthetic Antibody Phage-Display Libraries},
author = {Johan Nilvebrant and Sachdev S Sidhu},
doi = {10.1007/978-1-4939-7447-4_3},
issn = {1940-6029},
year = {2018},
date = {2018-01-01},
urldate = {2018-01-01},
journal = {Methods Mol Biol},
volume = {1701},
pages = {45--60},
abstract = {Synthetic antibody libraries provide a vast resource of renewable antibody reagents that can rival or exceed those of natural antibodies and can be rapidly isolated through controlled in vitro selections. Use of highly optimized human frameworks enables the incorporation of defined diversity at positions that are most likely to contribute to antigen recognition. This protocol describes the construction of synthetic antibody libraries based on a single engineered human autonomous variable heavy domain scaffold with diversity in all three complementarity-determining regions. The resulting libraries can be used to generate recombinant domain antibodies for a wide range of protein antigens using phage display. Furthermore, analogous methods can be used to construct antibody libraries based on larger antibody fragments or second-generation libraries aimed to fine-tune antibody characteristics including affinity, specificity, and manufacturability. The procedures rely on standard reagents and equipment available in most molecular biology laboratories.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
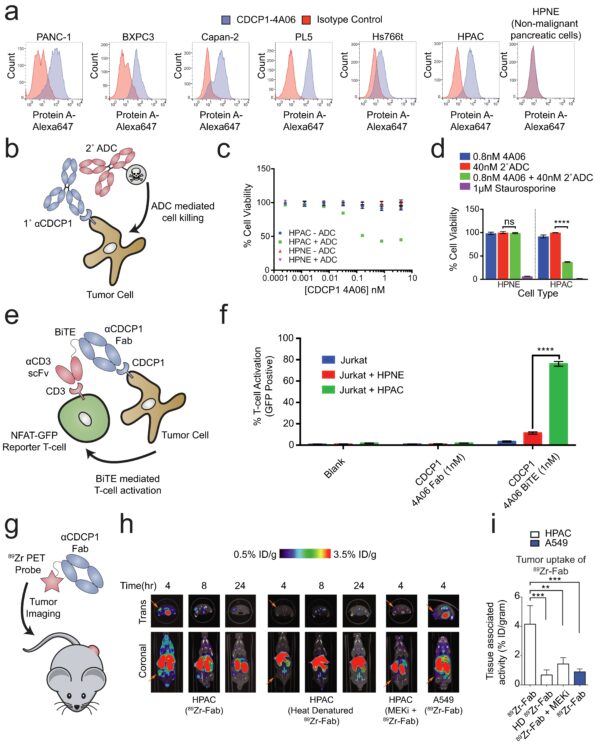
Martinko, Alexander J; Truillet, Charles; Julien, Olivier; Diaz, Juan E; Horlbeck, Max A; Whiteley, Gordon; Blonder, Josip; Weissman, Jonathan S; Bandyopadhyay, Sourav; Evans, Michael J; Wells, James A
Targeting RAS-driven human cancer cells with antibodies to upregulated and essential cell-surface proteins Journal Article
In: Elife, vol. 7, 2018, ISSN: 2050-084X.
@article{pmid29359686,
title = {Targeting RAS-driven human cancer cells with antibodies to upregulated and essential cell-surface proteins},
author = {Alexander J Martinko and Charles Truillet and Olivier Julien and Juan E Diaz and Max A Horlbeck and Gordon Whiteley and Josip Blonder and Jonathan S Weissman and Sourav Bandyopadhyay and Michael J Evans and James A Wells},
doi = {10.7554/eLife.31098},
issn = {2050-084X},
year = {2018},
date = {2018-01-01},
urldate = {2018-01-01},
journal = {Elife},
volume = {7},
abstract = {While there have been tremendous efforts to target oncogenic RAS signaling from inside the cell, little effort has focused on the cell-surface. Here, we used quantitative surface proteomics to reveal a signature of proteins that are upregulated on cells transformed with KRAS, and driven by MAPK pathway signaling. We next generated a toolkit of recombinant antibodies to seven of these RAS-induced proteins. We found that five of these proteins are broadly distributed on cancer cell lines harboring RAS mutations. In parallel, a cell-surface CRISPRi screen identified integrin and Wnt signaling proteins as critical to RAS-transformed cells. We show that antibodies targeting CDCP1, a protein common to our proteomics and CRISPRi datasets, can be leveraged to deliver cytotoxic and immunotherapeutic payloads to RAS-transformed cancer cells and report for RAS signaling status in vivo. Taken together, this work presents a technological platform for attacking RAS from outside the cell.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Weeks, Amy M; Wells, James A
Engineering peptide ligase specificity by proteomic identification of ligation sites Journal Article
In: Nat Chem Biol, vol. 14, no. 1, pp. 50–57, 2018, ISSN: 1552-4469.
@article{pmid29155430,
title = {Engineering peptide ligase specificity by proteomic identification of ligation sites},
author = {Amy M Weeks and James A Wells},
doi = {10.1038/nchembio.2521},
issn = {1552-4469},
year = {2018},
date = {2018-01-01},
urldate = {2018-01-01},
journal = {Nat Chem Biol},
volume = {14},
number = {1},
pages = {50--57},
abstract = {Enzyme-catalyzed peptide ligation is a powerful tool for site-specific protein bioconjugation, but stringent enzyme-substrate specificity limits its utility. We developed an approach for comprehensively characterizing peptide ligase specificity for N termini using proteome-derived peptide libraries. We used this strategy to characterize the ligation efficiency for >25,000 enzyme-substrate pairs in the context of the engineered peptide ligase subtiligase and identified a family of 72 mutant subtiligases with activity toward N-terminal sequences that were previously recalcitrant to modification. We applied these mutants individually for site-specific bioconjugation of purified proteins, including antibodies, and in algorithmically selected combinations for sequencing of the cellular N terminome with reduced sequence bias. We also developed a web application to enable algorithmic selection of the most efficient subtiligase variant(s) for bioconjugation to user-defined sequences. Our methods provide a new toolbox of enzymes for site-specific protein modification and a general approach for rapidly defining and engineering peptide ligase specificity.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Srinivasan, Sharan R; Cesa, Laura C; Li, Xiaokai; Julien, Olivier; Zhuang, Min; Shao, Hao; Chung, Jooho; Maillard, Ivan; Wells, James A; Duckett, Colin S; Gestwicki, Jason E
Heat Shock Protein 70 (Hsp70) Suppresses RIP1-Dependent Apoptotic and Necroptotic Cascades Journal Article
In: Mol Cancer Res, vol. 16, no. 1, pp. 58–68, 2018, ISSN: 1557-3125.
@article{pmid28970360,
title = {Heat Shock Protein 70 (Hsp70) Suppresses RIP1-Dependent Apoptotic and Necroptotic Cascades},
author = {Sharan R Srinivasan and Laura C Cesa and Xiaokai Li and Olivier Julien and Min Zhuang and Hao Shao and Jooho Chung and Ivan Maillard and James A Wells and Colin S Duckett and Jason E Gestwicki},
doi = {10.1158/1541-7786.MCR-17-0408},
issn = {1557-3125},
year = {2018},
date = {2018-01-01},
urldate = {2018-01-01},
journal = {Mol Cancer Res},
volume = {16},
number = {1},
pages = {58--68},
abstract = {Hsp70 is a molecular chaperone that binds to "client" proteins and protects them from protein degradation. Hsp70 is essential for the survival of many cancer cells, but it is not yet clear which of its clients are involved. Using structurally distinct chemical inhibitors, we found that many of the well-known clients of the related chaperone, Hsp90, are not strikingly responsive to Hsp70 inhibition. Rather, Hsp70 appeared to be important for the stability of the RIP1 (RIPK1) regulators: cIAP1/2 (BIRC1 and BIRC3), XIAP, and cFLIP (CFLAR). These results suggest that Hsp70 limits apoptosis and necroptosis pathways downstream of RIP1. Consistent with this model, MDA-MB-231 breast cancer cells treated with Hsp70 inhibitors underwent apoptosis, while cotreatment with z-VAD.fmk switched the cell death pathway to necroptosis. In addition, cell death in response to Hsp70 inhibitors was strongly suppressed by RIP1 knockdown or inhibitors. Thus, these data indicate that Hsp70 plays a previously unrecognized and important role in suppressing RIP1 activity. These findings clarify the role of Hsp70 in prosurvival signaling and suggest IAPs as potential new biomarkers for Hsp70 inhibition. .},
keywords = {},
pubstate = {published},
tppubtype = {article}
}