Publications: 2016
2016
Spangler, Benjamin; Fontaine, Shaun D; Shi, Yihui; Sambucetti, Lidia; Mattis, Aras N; Hann, Byron; Wells, James A; Renslo, Adam R
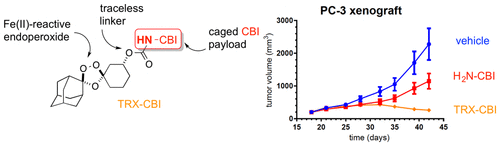
A Novel Tumor-Activated Prodrug Strategy Targeting Ferrous Iron Is Effective in Multiple Preclinical Cancer Models Journal Article
In: J Med Chem, vol. 59, no. 24, pp. 11161–11170, 2016, ISSN: 1520-4804.
@article{pmid27936709,
title = {A Novel Tumor-Activated Prodrug Strategy Targeting Ferrous Iron Is Effective in Multiple Preclinical Cancer Models},
author = {Benjamin Spangler and Shaun D Fontaine and Yihui Shi and Lidia Sambucetti and Aras N Mattis and Byron Hann and James A Wells and Adam R Renslo},
doi = {10.1021/acs.jmedchem.6b01470},
issn = {1520-4804},
year = {2016},
date = {2016-12-01},
urldate = {2016-12-01},
journal = {J Med Chem},
volume = {59},
number = {24},
pages = {11161--11170},
abstract = {Here we describe a new approach for tumor targeting in which augmented concentrations of Fe(II) in cancer cells and/or the tumor microenvironment triggers drug release from an Fe(II)-reactive prodrug conjugate. The 1,2,4-trioxolane scaffold developed to enable this approach can in principle be applied to a broad range of cancer therapeutics and is illustrated here with Fe(II)-targeted forms of a microtubule toxin and a duocarmycin-class DNA-alkylating agent. We show that the intrinsic reactivity/toxicity of the duocarmycin analog is masked in the conjugated form and this greatly reduced toxicity in mice. This in turn permitted elevated dosing levels, leading to higher systemic exposure and a significantly improved response in tumor xenograft models. Overall our results suggest that Fe(II)-dependent drug delivery via trioxolane conjugates could have significant utility in expanding the therapeutic index of a range of clinical and preclinical stage cancer chemotherapeutics.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Ye, Xiaoying; Chan, King C; Waters, Andrew M; Bess, Matthew; Harned, Adam; Wei, Bih-Rong; Loncarek, Jadranka; Luke, Brian T; Orsburn, Benjamin C; Hollinger, Bradley D; Stephens, Robert M; Bagni, Rachel; Martinko, Alex; Wells, James A; Nissley, Dwight V; McCormick, Frank; Whiteley, Gordon; Blonder, Josip
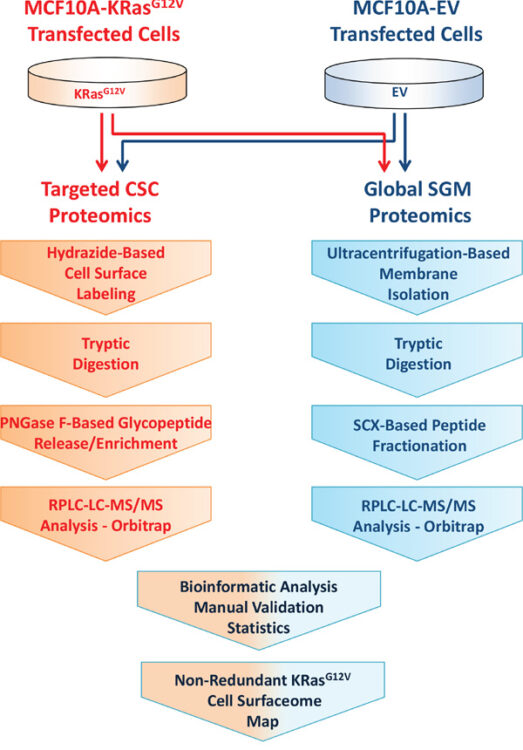
Comparative proteomics of a model MCF10A-KRasG12V cell line reveals a distinct molecular signature of the KRasG12V cell surface Journal Article
In: Oncotarget, vol. 7, no. 52, pp. 86948–86971, 2016, ISSN: 1949-2553.
@article{pmid27894102,
title = {Comparative proteomics of a model MCF10A-KRasG12V cell line reveals a distinct molecular signature of the KRasG12V cell surface},
author = {Xiaoying Ye and King C Chan and Andrew M Waters and Matthew Bess and Adam Harned and Bih-Rong Wei and Jadranka Loncarek and Brian T Luke and Benjamin C Orsburn and Bradley D Hollinger and Robert M Stephens and Rachel Bagni and Alex Martinko and James A Wells and Dwight V Nissley and Frank McCormick and Gordon Whiteley and Josip Blonder},
doi = {10.18632/oncotarget.13566},
issn = {1949-2553},
year = {2016},
date = {2016-12-01},
urldate = {2016-12-01},
journal = {Oncotarget},
volume = {7},
number = {52},
pages = {86948--86971},
abstract = {Oncogenic Ras mutants play a major role in the etiology of most aggressive and deadly carcinomas in humans. In spite of continuous efforts, effective pharmacological treatments targeting oncogenic Ras isoforms have not been developed. Cell-surface proteins represent top therapeutic targets primarily due to their accessibility and susceptibility to different modes of cancer therapy. To expand the treatment options of cancers driven by oncogenic Ras, new targets need to be identified and characterized at the surface of cancer cells expressing oncogenic Ras mutants. Here, we describe a mass spectrometry-based method for molecular profiling of the cell surface using KRasG12V transfected MCF10A (MCF10A-KRasG12V) as a model cell line of constitutively activated KRas and native MCF10A cells transduced with an empty vector (EV) as control. An extensive molecular map of the KRas surface was achieved by applying, in parallel, targeted hydrazide-based cell-surface capturing technology and global shotgun membrane proteomics to identify the proteins on the KRasG12V surface. This method allowed for integrated proteomic analysis that identified more than 500 cell-surface proteins found unique or upregulated on the surface of MCF10A-KRasG12V cells. Multistep bioinformatic processing was employed to elucidate and prioritize targets for cross-validation. Scanning electron microscopy and phenotypic cancer cell assays revealed changes at the cell surface consistent with malignant epithelial-to-mesenchymal transformation secondary to KRasG12V activation. Taken together, this dataset significantly expands the map of the KRasG12V surface and uncovers potential targets involved primarily in cell motility, cellular protrusion formation, and metastasis.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Pan, Yuchen; Sackmann, Eric K; Wypisniak, Karolina; Hornsby, Michael; Datwani, Sammy S; Herr, Amy E
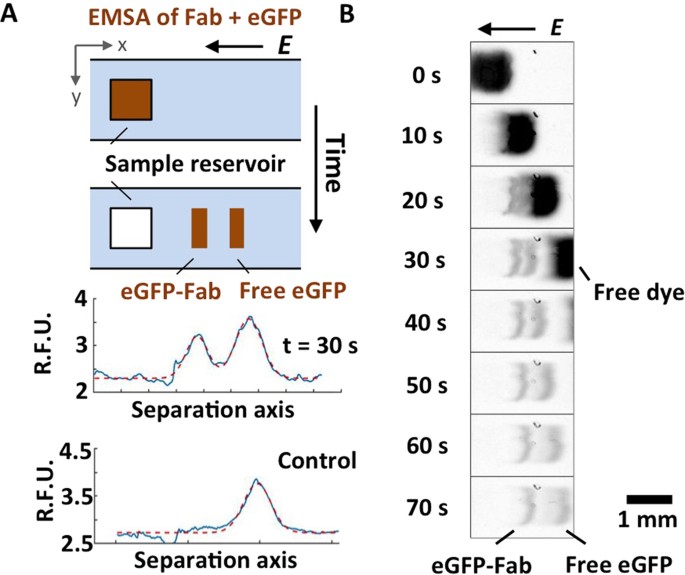
Determination of equilibrium dissociation constants for recombinant antibodies by high-throughput affinity electrophoresis Journal Article
In: Sci Rep, vol. 6, pp. 39774, 2016, ISSN: 2045-2322.
@article{pmid28008969,
title = {Determination of equilibrium dissociation constants for recombinant antibodies by high-throughput affinity electrophoresis},
author = {Yuchen Pan and Eric K Sackmann and Karolina Wypisniak and Michael Hornsby and Sammy S Datwani and Amy E Herr},
doi = {10.1038/srep39774},
issn = {2045-2322},
year = {2016},
date = {2016-12-01},
urldate = {2016-12-01},
journal = {Sci Rep},
volume = {6},
pages = {39774},
abstract = {High-quality immunoreagents enhance the performance and reproducibility of immunoassays and, in turn, the quality of both biological and clinical measurements. High quality recombinant immunoreagents are generated using antibody-phage display. One metric of antibody quality - the binding affinity - is quantified through the dissociation constant (K) of each recombinant antibody and the target antigen. To characterize the K of recombinant antibodies and target antigen, we introduce affinity electrophoretic mobility shift assays (EMSAs) in a high-throughput format suitable for small volume samples. A microfluidic card comprised of free-standing polyacrylamide gel (fsPAG) separation lanes supports 384 concurrent EMSAs in 30 s using a single power source. Sample is dispensed onto the microfluidic EMSA card by acoustic droplet ejection (ADE), which reduces EMSA variability compared to sample dispensing using manual or pin tools. The K for each of a six-member fragment antigen-binding fragment library is reported using ~25-fold less sample mass and ~5-fold less time than conventional heterogeneous assays. Given the form factor and performance of this micro- and mesofluidic workflow, we have developed a sample-sparing, high-throughput, solution-phase alternative for biomolecular affinity characterization.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Henager, Samuel H; Chu, Nam; Chen, Zan; Bolduc, David; Dempsey, Daniel R; Hwang, Yousang; Wells, James; Cole, Philip A
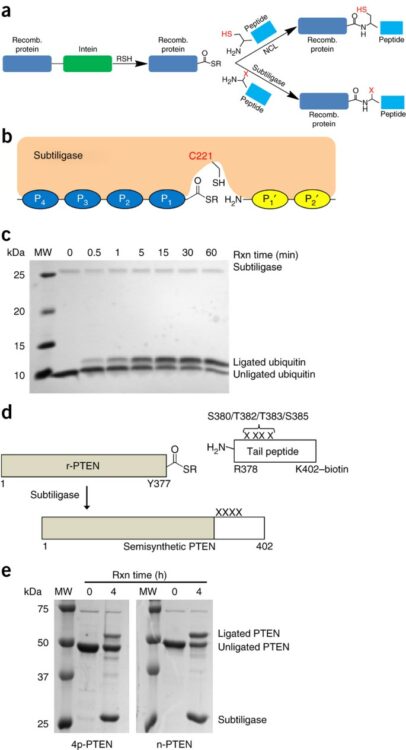
Enzyme-catalyzed expressed protein ligation Journal Article
In: Nat Methods, vol. 13, no. 11, pp. 925–927, 2016, ISSN: 1548-7105.
@article{pmid27669326,
title = {Enzyme-catalyzed expressed protein ligation},
author = {Samuel H Henager and Nam Chu and Zan Chen and David Bolduc and Daniel R Dempsey and Yousang Hwang and James Wells and Philip A Cole},
doi = {10.1038/nmeth.4004},
issn = {1548-7105},
year = {2016},
date = {2016-11-01},
urldate = {2016-11-01},
journal = {Nat Methods},
volume = {13},
number = {11},
pages = {925--927},
abstract = {Expressed protein ligation is a valuable method for protein semisynthesis that involves the reaction of recombinant protein C-terminal thioesters with N-terminal cysteine (N-Cys)-containing peptides, but the requirement of a Cys residue at the ligation junction can limit the utility of this method. Here we employ subtiligase variants to efficiently ligate Cys-free peptides to protein thioesters. Using this method, we have more accurately determined the effect of C-terminal phosphorylation on the tumor suppressor protein PTEN.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
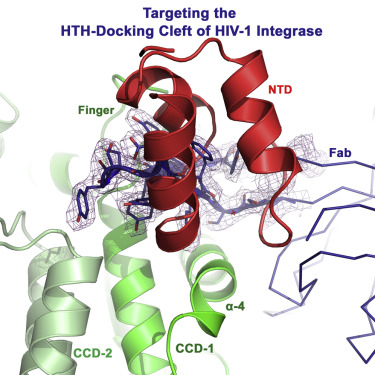
Galilee, Meytal; Britan-Rosich, Elena; Griner, Sarah L; Uysal, Serdar; Baumgärtel, Viola; Lamb, Don C; Kossiakoff, Anthony A; Kotler, Moshe; Stroud, Robert M; Marx, Ailie; Alian, Akram
The Preserved HTH-Docking Cleft of HIV-1 Integrase Is Functionally Critical Journal Article
In: Structure, vol. 24, no. 11, pp. 1936–1946, 2016, ISSN: 1878-4186.
@article{pmid27692964,
title = {The Preserved HTH-Docking Cleft of HIV-1 Integrase Is Functionally Critical},
author = {Meytal Galilee and Elena Britan-Rosich and Sarah L Griner and Serdar Uysal and Viola Baumgärtel and Don C Lamb and Anthony A Kossiakoff and Moshe Kotler and Robert M Stroud and Ailie Marx and Akram Alian},
doi = {10.1016/j.str.2016.08.015},
issn = {1878-4186},
year = {2016},
date = {2016-11-01},
urldate = {2016-11-01},
journal = {Structure},
volume = {24},
number = {11},
pages = {1936--1946},
abstract = {HIV-1 integrase (IN) catalyzes viral DNA integration into the host genome and facilitates multifunctional steps including virus particle maturation. Competency of IN to form multimeric assemblies is functionally critical, presenting an approach for anti-HIV strategies. Multimerization of IN depends on interactions between the distinct subunit domains and among the flanking protomers. Here, we elucidate an overlooked docking cleft of IN core domain that anchors the N-terminal helix-turn-helix (HTH) motif in a highly preserved and functionally critical configuration. Crystallographic structure of IN core domain in complex with Fab specifically targeting this cleft reveals a steric overlap that would inhibit HTH-docking, C-terminal domain contacts, DNA binding, and subsequent multimerization. While Fab inhibits in vitro IN integration activity, in vivo it abolishes virus particle production by specifically associating with preprocessed IN within Gag-Pol and interfering with early cytosolic Gag/Gag-Pol assemblies. The HTH-docking cleft may offer a fresh hotspot for future anti-HIV intervention strategies.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
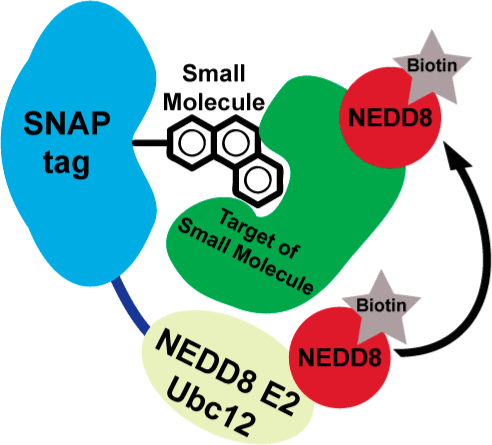
Hill, Zachary B; Pollock, Samuel B; Zhuang, Min; Wells, James A
Direct Proximity Tagging of Small Molecule Protein Targets Using an Engineered NEDD8 Ligase Journal Article
In: J Am Chem Soc, vol. 138, no. 40, pp. 13123–13126, 2016, ISSN: 1520-5126.
@article{pmid27626304,
title = {Direct Proximity Tagging of Small Molecule Protein Targets Using an Engineered NEDD8 Ligase},
author = {Zachary B Hill and Samuel B Pollock and Min Zhuang and James A Wells},
doi = {10.1021/jacs.6b06828},
issn = {1520-5126},
year = {2016},
date = {2016-10-01},
urldate = {2016-10-01},
journal = {J Am Chem Soc},
volume = {138},
number = {40},
pages = {13123--13126},
abstract = {Identifying the protein targets of bioactive small molecules remains a major problem in the discovery of new chemical probes and therapeutics. While activity-based probes and photo-cross-linkers have had success in identifying protein targets of small molecules, each technique has limitations. Here we describe a method for direct proximity tagging of proteins that bind small molecules. We engineered a promiscuous ligase based on the NEDD8 conjugating enzyme, Ubc12, which can be covalently linked to a small molecule of interest. When target proteins bind the small molecule, they are directly labeled on surface lysines with a biotinylated derivative of the small ubiquitin homologue, NEDD8. This unique covalent tag can then be used to identify the small molecule binding proteins. Utilizing the drug dasatinib, we have shown that dasatinib-directed NEDDylation occurs for known endogenous protein binders in complex cell lysates. In addition, we have been able to improve NEDDylation efficiency through rational mutagenesis. Finally, we have shown that affinity-directed NEDDylation can be applied to two other protein-ligand interactions beyond kinases. Proximity tagging using this engineered ligase requires direct binding of the target and, thus, provides a useful and orthogonal approach to facilitate small molecule target identification.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
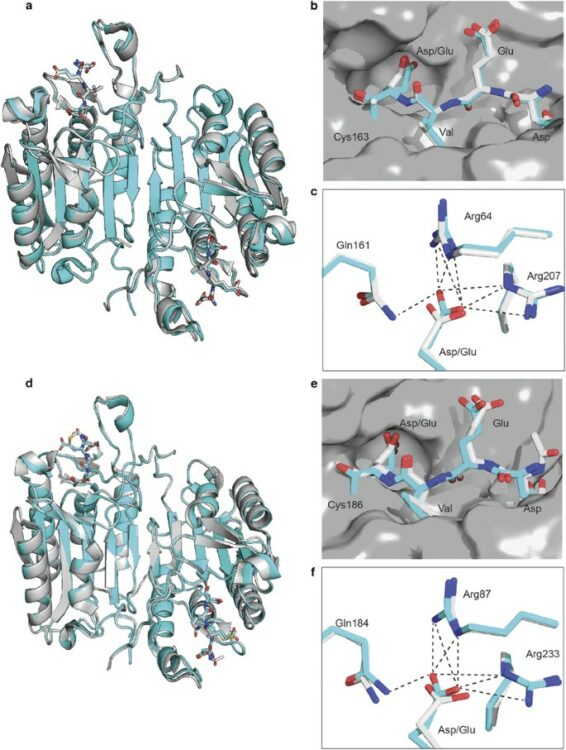
Seaman, J E; Julien, O; Lee, P S; Rettenmaier, T J; Thomsen, N D; Wells, J A
Cacidases: caspases can cleave after aspartate, glutamate and phosphoserine residues Journal Article
In: Cell Death Differ, vol. 23, no. 10, pp. 1717–1726, 2016, ISSN: 1476-5403.
@article{pmid27367566,
title = {Cacidases: caspases can cleave after aspartate, glutamate and phosphoserine residues},
author = {J E Seaman and O Julien and P S Lee and T J Rettenmaier and N D Thomsen and J A Wells},
doi = {10.1038/cdd.2016.62},
issn = {1476-5403},
year = {2016},
date = {2016-10-01},
urldate = {2016-10-01},
journal = {Cell Death Differ},
volume = {23},
number = {10},
pages = {1717--1726},
abstract = {Caspases are a family of proteases found in all metazoans, including a dozen in humans, that drive the terminal stages of apoptosis as well as other cellular remodeling and inflammatory events. Caspases are named because they are cysteine class enzymes shown to cleave after aspartate residues. In the past decade, we and others have developed unbiased proteomic methods that collectively identified ~2000 native proteins cleaved during apoptosis after the signature aspartate residues. Here, we explore non-aspartate cleavage events and identify 100s of substrates cleaved after glutamate in both human and murine apoptotic samples. The extended consensus sequence patterns are virtually identical for the aspartate and glutamate cleavage sites suggesting they are cleaved by the same caspases. Detailed kinetic analyses of the dominant apoptotic executioner caspases-3 and -7 show that synthetic substrates containing DEVD↓ are cleaved only twofold faster than DEVE↓, which is well within the 500-fold range of rates that natural proteins are cut. X-ray crystallography studies confirm that the two acidic substrates bind in virtually the same way to either caspases-3 or -7 with minimal adjustments to accommodate the larger glutamate. Lastly, during apoptosis we found 121 proteins cleaved after serine residues that have been previously annotated to be phosphorylation sites. We found that caspase-3, but not caspase-7, can cleave peptides containing DEVpS↓ at only threefold slower rate than DEVD↓, but does not cleave the unphosphorylated serine peptide. There are only a handful of previously reported examples of proteins cleaved after glutamate and none after phosphorserine. Our studies reveal a much greater promiscuity for cleaving after acidic residues and the name 'cacidase' could aptly reflect this broader specificity.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
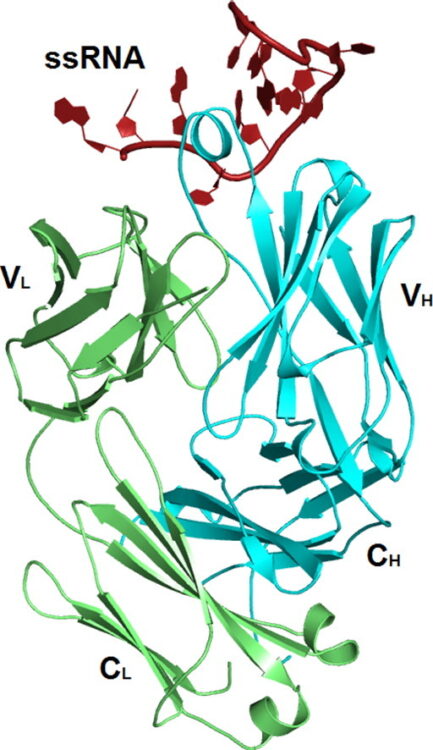
Shao, Yaming; Huang, Hao; Qin, Daoming; Li, Nan-Sheng; Koide, Akiko; Staley, Jonathan P; Koide, Shohei; Kossiakoff, Anthony A; Piccirilli, Joseph A
Specific Recognition of a Single-Stranded RNA Sequence by a Synthetic Antibody Fragment Journal Article
In: J Mol Biol, vol. 428, no. 20, pp. 4100–4114, 2016, ISSN: 1089-8638.
@article{pmid27593161,
title = {Specific Recognition of a Single-Stranded RNA Sequence by a Synthetic Antibody Fragment},
author = {Yaming Shao and Hao Huang and Daoming Qin and Nan-Sheng Li and Akiko Koide and Jonathan P Staley and Shohei Koide and Anthony A Kossiakoff and Joseph A Piccirilli},
doi = {10.1016/j.jmb.2016.08.029},
issn = {1089-8638},
year = {2016},
date = {2016-10-01},
urldate = {2016-10-01},
journal = {J Mol Biol},
volume = {428},
number = {20},
pages = {4100--4114},
abstract = {Antibodies that bind RNA represent an unrealized source of reagents for synthetic biology and for characterizing cellular transcriptomes. However, facile access to RNA-binding antibodies requires the engineering of effective Fab libraries guided by the knowledge of the principles that govern RNA recognition. Here, we describe a Fab identified from a minimalist synthetic library during phage display against a branched RNA target. The Fab (BRG) binds with 20nM dissociation constant to a single-stranded RNA (ssRNA) sequence adjacent to the branch site and can block the action of debranchase enzyme. We report the crystal structure in complex with RNA target at 2.38Å. The Fab traps the RNA in a hairpin conformation that contains a 2-bp duplex capped by a tetraloop. The paratope surface consists of residues located in four complementarity-determining regions including a major contribution from H3, which adopts a helical structure that projects into a deep, wide groove formed by the RNA. The amino acid composition of the paratope reflects the library diversity, consisting mostly of tyrosine and serine residues and a small but significant contribution from a single arginine residue. This structure, involving the recognition of ssRNA via a stem-loop conformation, together with our two previous structures involving the recognition of an RNA hairpin loop and an RNA tertiary structure, reveals the capacity of minimalist libraries biased with tyrosine, serine, glycine, and arginine to form binding surfaces for specific RNA conformations and distinct levels of RNA structural hierarchy.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
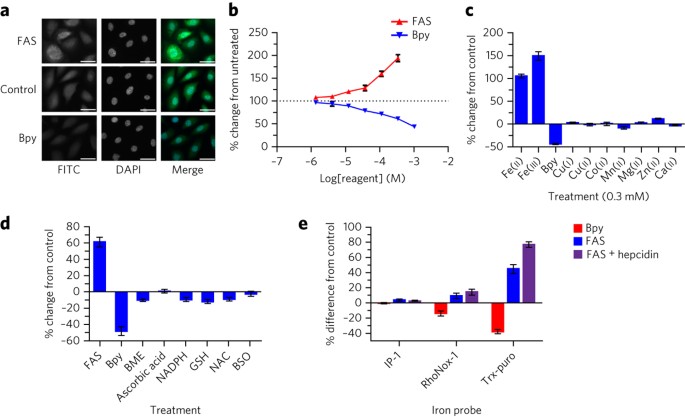
Spangler, Benjamin; Morgan, Charles W; Fontaine, Shaun D; Wal, Mark N Vander; Chang, Christopher J; Wells, James A; Renslo, Adam R
A reactivity-based probe of the intracellular labile ferrous iron pool Journal Article
In: Nat Chem Biol, vol. 12, no. 9, pp. 680–685, 2016, ISSN: 1552-4469.
@article{pmid27376690,
title = {A reactivity-based probe of the intracellular labile ferrous iron pool},
author = {Benjamin Spangler and Charles W Morgan and Shaun D Fontaine and Mark N Vander Wal and Christopher J Chang and James A Wells and Adam R Renslo},
doi = {10.1038/nchembio.2116},
issn = {1552-4469},
year = {2016},
date = {2016-09-01},
urldate = {2016-09-01},
journal = {Nat Chem Biol},
volume = {12},
number = {9},
pages = {680--685},
abstract = {Improved methods for studying intracellular reactive Fe(II) are of significant interest for studies of iron metabolism and disease-relevant changes in iron homeostasis. Here we describe a highly selective reactivity-based probe in which a Fenton-type reaction with intracellular labile Fe(II) leads to unmasking of the aminonucleoside puromycin. Puromycin leaves a permanent and dose-dependent mark on treated cells that can be detected with high sensitivity and precision using a high-content, plate-based immunofluorescence assay. Using this new probe and screening approach, we detected alteration of cellular labile Fe(II) in response extracellular iron conditioning, overexpression of iron storage and/or export proteins, and post-translational regulation of iron export. We also used this new tool to demonstrate that labile Fe(II) pools are larger in cancer cells than in nontumorigenic cells.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Wang, Wei; Nguyen, Linh T T; Burlak, Christopher; Chegini, Fariba; Guo, Feng; Chataway, Tim; Ju, Shulin; Fisher, Oriana S; Miller, David W; Datta, Debajyoti; Wu, Fang; Wu, Chun-Xiang; Landeru, Anuradha; Wells, James A; Cookson, Mark R; Boxer, Matthew B; Thomas, Craig J; Gai, Wei Ping; Ringe, Dagmar; Petsko, Gregory A; Hoang, Quyen Q
Caspase-1 causes truncation and aggregation of the Parkinson's disease-associated protein α-synuclein Journal Article
In: Proc Natl Acad Sci U S A, vol. 113, no. 34, pp. 9587–9592, 2016, ISSN: 1091-6490.
@article{pmid27482083,
title = {Caspase-1 causes truncation and aggregation of the Parkinson's disease-associated protein α-synuclein},
author = {Wei Wang and Linh T T Nguyen and Christopher Burlak and Fariba Chegini and Feng Guo and Tim Chataway and Shulin Ju and Oriana S Fisher and David W Miller and Debajyoti Datta and Fang Wu and Chun-Xiang Wu and Anuradha Landeru and James A Wells and Mark R Cookson and Matthew B Boxer and Craig J Thomas and Wei Ping Gai and Dagmar Ringe and Gregory A Petsko and Quyen Q Hoang},
doi = {10.1073/pnas.1610099113},
issn = {1091-6490},
year = {2016},
date = {2016-08-01},
urldate = {2016-08-01},
journal = {Proc Natl Acad Sci U S A},
volume = {113},
number = {34},
pages = {9587--9592},
abstract = {The aggregation of α-synuclein (aSyn) leading to the formation of Lewy bodies is the defining pathological hallmark of Parkinson's disease (PD). Rare familial PD-associated mutations in aSyn render it aggregation-prone; however, PD patients carrying wild type (WT) aSyn also have aggregated aSyn in Lewy bodies. The mechanisms by which WT aSyn aggregates are unclear. Here, we report that inflammation can play a role in causing the aggregation of WT aSyn. We show that activation of the inflammasome with known stimuli results in the aggregation of aSyn in a neuronal cell model of PD. The insoluble aggregates are enriched with truncated aSyn as found in Lewy bodies of the PD brain. Inhibition of the inflammasome enzyme caspase-1 by chemical inhibition or genetic knockdown with shRNA abated aSyn truncation. In vitro characterization confirmed that caspase-1 directly cleaves aSyn, generating a highly aggregation-prone species. The truncation-induced aggregation of aSyn is toxic to neuronal culture, and inhibition of caspase-1 by shRNA or a specific chemical inhibitor improved the survival of a neuronal PD cell model. This study provides a molecular link for the role of inflammation in aSyn aggregation, and perhaps in the pathogenesis of sporadic PD as well.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Leung, Isabel; Dekel, Ayelet; Shifman, Julia M; Sidhu, Sachdev S
Saturation scanning of ubiquitin variants reveals a common hot spot for binding to USP2 and USP21 Journal Article
In: Proc Natl Acad Sci U S A, vol. 113, no. 31, pp. 8705–8710, 2016, ISSN: 1091-6490.
@article{pmid27436899,
title = {Saturation scanning of ubiquitin variants reveals a common hot spot for binding to USP2 and USP21},
author = {Isabel Leung and Ayelet Dekel and Julia M Shifman and Sachdev S Sidhu},
doi = {10.1073/pnas.1524648113},
issn = {1091-6490},
year = {2016},
date = {2016-08-01},
urldate = {2016-08-01},
journal = {Proc Natl Acad Sci U S A},
volume = {113},
number = {31},
pages = {8705--8710},
abstract = {A detailed understanding of the molecular mechanisms whereby ubiquitin (Ub) recognizes enzymes in the Ub proteasome system is crucial for understanding the biological function of Ub. Many structures of Ub complexes have been solved and, in most cases, reveal a large structural epitope on a common face of the Ub molecule. However, owing to the generally weak nature of these interactions, it has been difficult to map in detail the functional contributions of individual Ub side chains to affinity and specificity. Here we took advantage of Ub variants (Ubvs) that bind tightly to particular Ub-specific proteases (USPs) and used phage display and saturation scanning mutagenesis to comprehensively map functional epitopes within the structural epitopes. We found that Ubvs that bind to USP2 or USP21 contain a remarkably similar core functional epitope, or "hot spot," consisting mainly of positions that are conserved as the wild type sequence, but also some positions that prefer mutant sequences. The Ubv core functional epitope contacts residues that are conserved in the human USP family, and thus it is likely important for the interactions of Ub across many family members.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
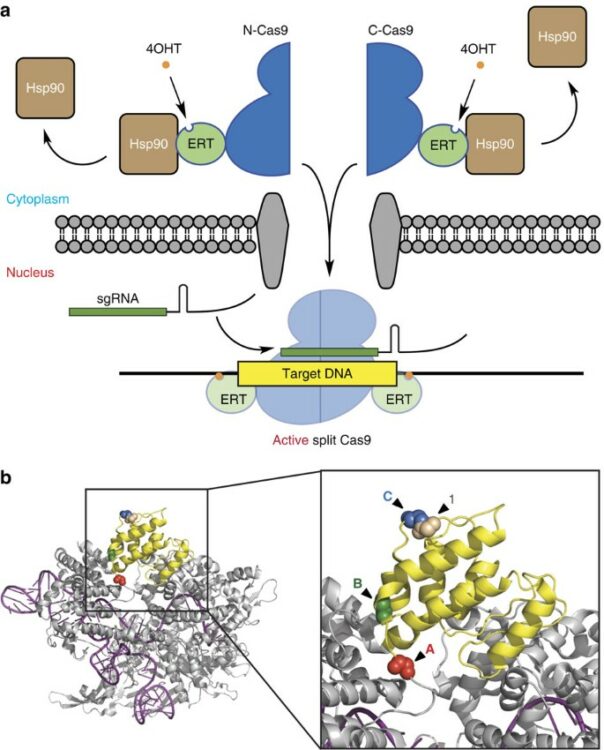
Nguyen, Duy P; Miyaoka, Yuichiro; Gilbert, Luke A; Mayerl, Steven J; Lee, Brian H; Weissman, Jonathan S; Conklin, Bruce R; Wells, James A
Ligand-binding domains of nuclear receptors facilitate tight control of split CRISPR activity Journal Article
In: Nat Commun, vol. 7, pp. 12009, 2016, ISSN: 2041-1723.
@article{pmid27363581,
title = {Ligand-binding domains of nuclear receptors facilitate tight control of split CRISPR activity},
author = {Duy P Nguyen and Yuichiro Miyaoka and Luke A Gilbert and Steven J Mayerl and Brian H Lee and Jonathan S Weissman and Bruce R Conklin and James A Wells},
doi = {10.1038/ncomms12009},
issn = {2041-1723},
year = {2016},
date = {2016-07-01},
urldate = {2016-07-01},
journal = {Nat Commun},
volume = {7},
pages = {12009},
abstract = {Cas9-based RNA-guided nuclease (RGN) has emerged to be a versatile method for genome editing due to the ease of construction of RGN reagents to target specific genomic sequences. The ability to control the activity of Cas9 with a high temporal resolution will facilitate tight regulation of genome editing processes for studying the dynamics of transcriptional regulation or epigenetic modifications in complex biological systems. Here we show that fusing ligand-binding domains of nuclear receptors to split Cas9 protein fragments can provide chemical control over split Cas9 activity. The method has allowed us to control Cas9 activity in a tunable manner with no significant background, which has been challenging for other inducible Cas9 constructs. We anticipate that our design will provide opportunities through the use of different ligand-binding domains to enable multiplexed genome regulation of endogenous genes in distinct loci through simultaneous chemical regulation of orthogonal Cas9 variants.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Schaefer, Zachary P; Bailey, Lucas J; Kossiakoff, Anthony A
A polar ring endows improved specificity to an antibody fragment Journal Article
In: Protein Sci, vol. 25, no. 7, pp. 1290–1298, 2016, ISSN: 1469-896X.
@article{pmid27334407,
title = {A polar ring endows improved specificity to an antibody fragment},
author = {Zachary P Schaefer and Lucas J Bailey and Anthony A Kossiakoff},
doi = {10.1002/pro.2888},
issn = {1469-896X},
year = {2016},
date = {2016-07-01},
urldate = {2016-07-01},
journal = {Protein Sci},
volume = {25},
number = {7},
pages = {1290--1298},
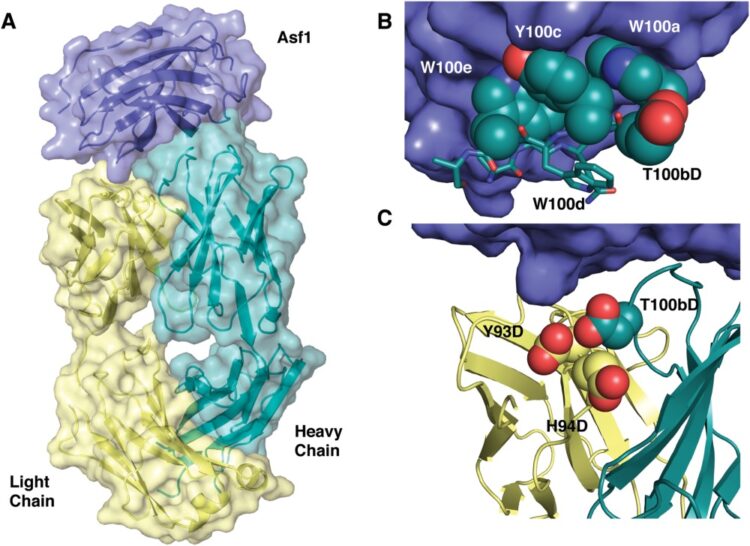
abstract = {Engineering monovalent Fab fragments into bivalent formats like IgGs or F(ab')2 can lead to aggregation presumably because of nonspecific off-target interactions that induce aggregation. In an effort to further understand the molecular determinants of nonspecific interactions for engineered antibodies and natively folded proteins in general, we focused on a synthetic Fab with low nanomolar affinity to histone chaperone Anti-silencing factor 1 (Asf1) that demonstrates off-target binding through low solubility (∼5 mg/mL) in the multivalent F(ab') 2 state. Here, we generated phage display-based shotgun scanning libraries to introduce aspartate as a negative design element into the antibody paratope. The antibody-combining site was amenable to aspartate substitution at numerous positions within the antigen binding loops and one variant, Tyr(L93) Asp/His(L94) Asp/Thr(H100b) Asp, possessed high solubility (>100 mg/ml). Furthermore, the mutations decreased nonspecific interactions measured by column interaction chromatography and ELISA in the multivalent antibody format while maintaining high affinity to the antigen. Structural determination of the antibody-antigen complex revealed that the aspartate-permissive residues formed a polar ring around the structural and functional paratope, recapitulating the canonical feature of naturally occurring protein-protein interactions. This observation may inform future strategies for the design and engineering of molecular recognition.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Hill, Maureen E; MacPherson, Derek J; Wu, Peng; Julien, Olivier; Wells, James A; Hardy, Jeanne A
Reprogramming Caspase-7 Specificity by Regio-Specific Mutations and Selection Provides Alternate Solutions for Substrate Recognition Journal Article
In: ACS Chem Biol, vol. 11, no. 6, pp. 1603–1612, 2016, ISSN: 1554-8937.
@article{pmid27032039,
title = {Reprogramming Caspase-7 Specificity by Regio-Specific Mutations and Selection Provides Alternate Solutions for Substrate Recognition},
author = {Maureen E Hill and Derek J MacPherson and Peng Wu and Olivier Julien and James A Wells and Jeanne A Hardy},
doi = {10.1021/acschembio.5b00971},
issn = {1554-8937},
year = {2016},
date = {2016-06-01},
urldate = {2016-06-01},
journal = {ACS Chem Biol},
volume = {11},
number = {6},
pages = {1603--1612},
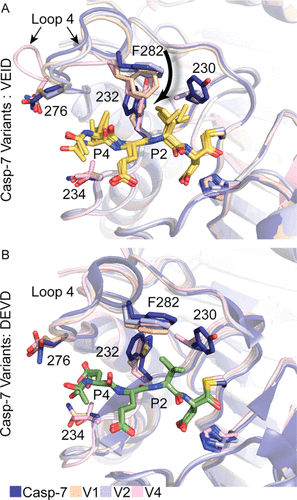
abstract = {The ability to routinely engineer protease specificity can allow us to better understand and modulate their biology for expanded therapeutic and industrial applications. Here, we report a new approach based on a caged green fluorescent protein (CA-GFP) reporter that allows for flow-cytometry-based selection in bacteria or other cell types enabling selection of intracellular protease specificity, regardless of the compositional complexity of the protease. Here, we apply this approach to introduce the specificity of caspase-6 into caspase-7, an intracellular cysteine protease important in cellular remodeling and cell death. We found that substitution of substrate-contacting residues from caspase-6 into caspase-7 was ineffective, yielding an inactive enzyme, whereas saturation mutagenesis at these positions and selection by directed evolution produced active caspases. The process produced a number of nonobvious mutations that enabled conversion of the caspase-7 specificity to match caspase-6. The structures of the evolved-specificity caspase-7 (esCasp-7) revealed alternate binding modes for the substrate, including reorganization of an active site loop. Profiling the entire human proteome of esCasp-7 by N-terminomics demonstrated that the global specificity toward natural protein substrates is remarkably similar to that of caspase-6. Because the esCasp-7 maintained the core of caspase-7, we were able to identify a caspase-6 substrate, lamin C, that we predict relies on an exosite for substrate recognition. These reprogrammed proteases may be the first tool built with the express intent of distinguishing exosite dependent or independent substrates. This approach to specificity reprogramming should also be generalizable across a wide range of proteases.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Arita, Yuko; Allen, Samantha; Chen, Gang; Zhang, Wei; Wang, Ying; Owen, Albert J; Dentinger, Patrick; Sidhu, Sachdev S
Rapid isolation of peptidic inhibitors of the solute carrier family transporters OATP1B1 and OATP1B3 by cell-based phage display selections Journal Article
In: Biochem Biophys Res Commun, vol. 473, no. 2, pp. 370–376, 2016, ISSN: 1090-2104.
@article{pmid26792727,
title = {Rapid isolation of peptidic inhibitors of the solute carrier family transporters OATP1B1 and OATP1B3 by cell-based phage display selections},
author = {Yuko Arita and Samantha Allen and Gang Chen and Wei Zhang and Ying Wang and Albert J Owen and Patrick Dentinger and Sachdev S Sidhu},
doi = {10.1016/j.bbrc.2016.01.050},
issn = {1090-2104},
year = {2016},
date = {2016-04-01},
urldate = {2016-04-01},
journal = {Biochem Biophys Res Commun},
volume = {473},
number = {2},
pages = {370--376},
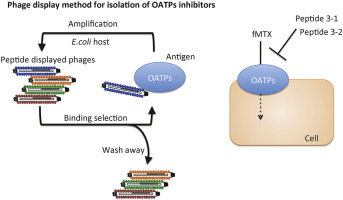
abstract = {OATP1B1 and OATP1B3 (1B3) are members of organic anion-transporting polypeptides (OATPs), a family of sodium-independent organic anion membrane transporters that contribute to transport of various drugs. To identify peptide inhibitors of OATP1B1, we developed a direct selection system on live cells using phage-displayed peptide libraries. Selections against OATP1B1 overexpressed cell-lines yielded three unique peptides able to inhibit the transport function of OATP1B1 and 1B3. Affinity maturation of one peptide led to identification of two peptides that demonstrated improved inhibition efficacy on drug uptake mediated by OATP1B1 and 1B3. We anticipate that these peptides will assist the identification of novel substrates for OATP1B1 and 1B3. Moreover, our selection system is a practical method for generating inhibitors of other membrane transporters.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Na, Hong; Laver, John D; Jeon, Jouhyun; Singh, Fateh; Ancevicius, Kristin; Fan, Yujie; Cao, Wen Xi; Nie, Kun; Yang, Zhenglin; Luo, Hua; Wang, Miranda; Rissland, Olivia; Westwood, J Timothy; Kim, Philip M; Smibert, Craig A; Lipshitz, Howard D; Sidhu, Sachdev S
A high-throughput pipeline for the production of synthetic antibodies for analysis of ribonucleoprotein complexes Journal Article
In: RNA, vol. 22, no. 4, pp. 636–655, 2016, ISSN: 1469-9001.
@article{pmid26847261,
title = {A high-throughput pipeline for the production of synthetic antibodies for analysis of ribonucleoprotein complexes},
author = {Hong Na and John D Laver and Jouhyun Jeon and Fateh Singh and Kristin Ancevicius and Yujie Fan and Wen Xi Cao and Kun Nie and Zhenglin Yang and Hua Luo and Miranda Wang and Olivia Rissland and J Timothy Westwood and Philip M Kim and Craig A Smibert and Howard D Lipshitz and Sachdev S Sidhu},
doi = {10.1261/rna.055186.115},
issn = {1469-9001},
year = {2016},
date = {2016-04-01},
journal = {RNA},
volume = {22},
number = {4},
pages = {636--655},
abstract = {Post-transcriptional regulation of mRNAs plays an essential role in the control of gene expression. mRNAs are regulated in ribonucleoprotein (RNP) complexes by RNA-binding proteins (RBPs) along with associated protein and noncoding RNA (ncRNA) cofactors. A global understanding of post-transcriptional control in any cell type requires identification of the components of all of its RNP complexes. We have previously shown that these complexes can be purified by immunoprecipitation using anti-RBP synthetic antibodies produced by phage display. To develop the large number of synthetic antibodies required for a global analysis of RNP complex composition, we have established a pipeline that combines (i) a computationally aided strategy for design of antigens located outside of annotated domains, (ii) high-throughput antigen expression and purification in Escherichia coli, and (iii) high-throughput antibody selection and screening. Using this pipeline, we have produced 279 antibodies against 61 different protein components of Drosophila melanogaster RNPs. Together with those produced in our low-throughput efforts, we have a panel of 311 antibodies for 67 RNP complex proteins. Tests of a subset of our antibodies demonstrated that 89% immunoprecipitate their endogenous target from embryo lysate. This panel of antibodies will serve as a resource for global studies of RNP complexes in Drosophila. Furthermore, our high-throughput pipeline permits efficient production of synthetic antibodies against any large set of proteins.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Julien, Olivier; Zhuang, Min; Wiita, Arun P; O'Donoghue, Anthony J; Knudsen, Giselle M; Craik, Charles S; Wells, James A
Quantitative MS-based enzymology of caspases reveals distinct protein substrate specificities, hierarchies, and cellular roles Journal Article
In: Proc Natl Acad Sci U S A, vol. 113, no. 14, pp. E2001–E2010, 2016, ISSN: 1091-6490.
@article{pmid27006500,
title = {Quantitative MS-based enzymology of caspases reveals distinct protein substrate specificities, hierarchies, and cellular roles},
author = {Olivier Julien and Min Zhuang and Arun P Wiita and Anthony J O'Donoghue and Giselle M Knudsen and Charles S Craik and James A Wells},
doi = {10.1073/pnas.1524900113},
issn = {1091-6490},
year = {2016},
date = {2016-04-01},
urldate = {2016-04-01},
journal = {Proc Natl Acad Sci U S A},
volume = {113},
number = {14},
pages = {E2001--E2010},
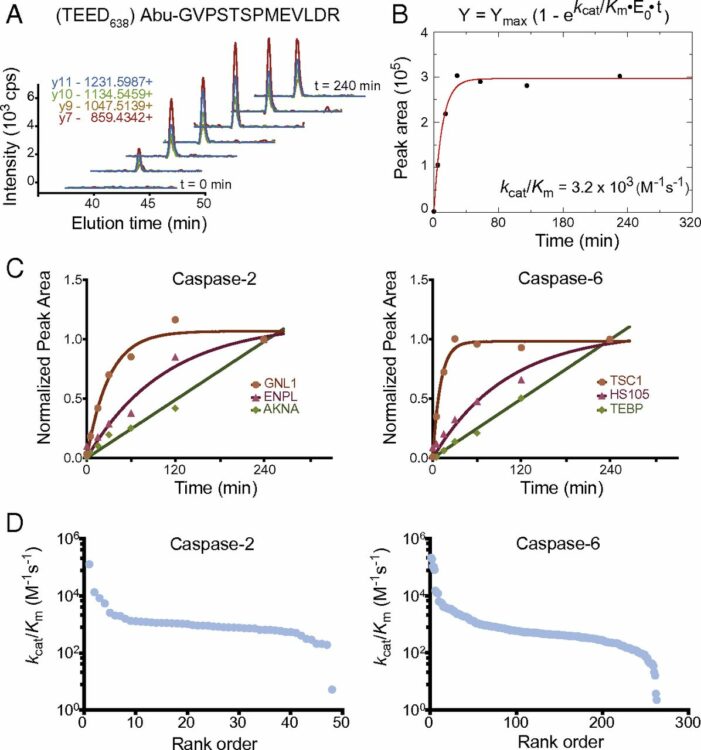
abstract = {Proteases constitute the largest enzyme family, yet their biological roles are obscured by our rudimentary understanding of their cellular substrates. There are 12 human caspases that play crucial roles in inflammation and cell differentiation and drive the terminal stages of cell death. Recent N-terminomics technologies have begun to enumerate the diverse substrates individual caspases can cleave in complex cell lysates. It is clear that many caspases have shared substrates; however, few data exist about the catalytic efficiencies (kcat/KM) of these substrates, which is critical to understanding their true substrate preferences. In this study, we use quantitative MS to determine the catalytic efficiencies for hundreds of natural protease substrates in cellular lysate for two understudied members: caspase-2 and caspase-6. Most substrates are new, and the cleavage rates vary up to 500-fold. We compare the cleavage rates for common substrates with those found for caspase-3, caspase-7, and caspase-8, involved in apoptosis. There is little correlation in catalytic efficiencies among the five caspases, suggesting each has a unique set of preferred substrates, and thus more specialized roles than previously understood. We synthesized peptide substrates on the basis of protein cleavage sites and found similar catalytic efficiencies between the protein and peptide substrates. These data suggest the rates of proteolysis are dominated more by local primary sequence, and less by the tertiary protein fold. Our studies highlight that global quantitative rate analysis for posttranslational modification enzymes in complex milieus for native substrates is critical to better define their functions and relative sequence of events.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Gorelik, Maryna; Orlicky, Stephen; Sartori, Maria A; Tang, Xiaojing; Marcon, Edyta; Kurinov, Igor; Greenblatt, Jack F; Tyers, Mike; Moffat, Jason; Sicheri, Frank; Sidhu, Sachdev S
Inhibition of SCF ubiquitin ligases by engineered ubiquitin variants that target the Cul1 binding site on the Skp1-F-box interface Journal Article
In: Proc Natl Acad Sci U S A, vol. 113, no. 13, pp. 3527–3532, 2016, ISSN: 1091-6490.
@article{pmid26976582,
title = {Inhibition of SCF ubiquitin ligases by engineered ubiquitin variants that target the Cul1 binding site on the Skp1-F-box interface},
author = {Maryna Gorelik and Stephen Orlicky and Maria A Sartori and Xiaojing Tang and Edyta Marcon and Igor Kurinov and Jack F Greenblatt and Mike Tyers and Jason Moffat and Frank Sicheri and Sachdev S Sidhu},
doi = {10.1073/pnas.1519389113},
issn = {1091-6490},
year = {2016},
date = {2016-03-01},
urldate = {2016-03-01},
journal = {Proc Natl Acad Sci U S A},
volume = {113},
number = {13},
pages = {3527--3532},
abstract = {Skp1-Cul1-F-box (SCF) E3 ligases play key roles in multiple cellular processes through ubiquitination and subsequent degradation of substrate proteins. Although Skp1 and Cul1 are invariant components of all SCF complexes, the 69 different human F-box proteins are variable substrate binding modules that determine specificity. SCF E3 ligases are activated in many cancers and inhibitors could have therapeutic potential. Here, we used phage display to develop specific ubiquitin-based inhibitors against two F-box proteins, Fbw7 and Fbw11. Unexpectedly, the ubiquitin variants bind at the interface of Skp1 and F-box proteins and inhibit ligase activity by preventing Cul1 binding to the same surface. Using structure-based design and phage display, we modified the initial inhibitors to generate broad-spectrum inhibitors that targeted many SCF ligases, or conversely, a highly specific inhibitor that discriminated between even the close homologs Fbw11 and Fbw1. We propose that most F-box proteins can be targeted by this approach for basic research and for potential cancer therapies.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
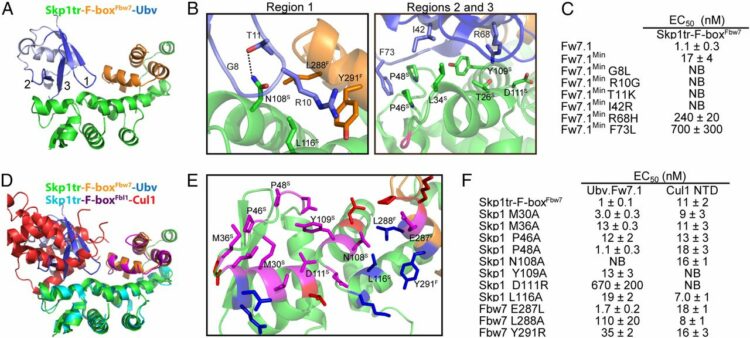
Cimermancic, Peter; Weinkam, Patrick; Rettenmaier, T Justin; Bichmann, Leon; Keedy, Daniel A; Woldeyes, Rahel A; Schneidman-Duhovny, Dina; Demerdash, Omar N; Mitchell, Julie C; Wells, James A; Fraser, James S; Sali, Andrej
CryptoSite: Expanding the Druggable Proteome by Characterization and Prediction of Cryptic Binding Sites Journal Article
In: J Mol Biol, vol. 428, no. 4, pp. 709–719, 2016, ISSN: 1089-8638.
@article{pmid26854760,
title = {CryptoSite: Expanding the Druggable Proteome by Characterization and Prediction of Cryptic Binding Sites},
author = {Peter Cimermancic and Patrick Weinkam and T Justin Rettenmaier and Leon Bichmann and Daniel A Keedy and Rahel A Woldeyes and Dina Schneidman-Duhovny and Omar N Demerdash and Julie C Mitchell and James A Wells and James S Fraser and Andrej Sali},
doi = {10.1016/j.jmb.2016.01.029},
issn = {1089-8638},
year = {2016},
date = {2016-02-01},
urldate = {2016-02-01},
journal = {J Mol Biol},
volume = {428},
number = {4},
pages = {709--719},
abstract = {Many proteins have small-molecule binding pockets that are not easily detectable in the ligand-free structures. These cryptic sites require a conformational change to become apparent; a cryptic site can therefore be defined as a site that forms a pocket in a holo structure, but not in the apo structure. Because many proteins appear to lack druggable pockets, understanding and accurately identifying cryptic sites could expand the set of drug targets. Previously, cryptic sites were identified experimentally by fragment-based ligand discovery and computationally by long molecular dynamics simulations and fragment docking. Here, we begin by constructing a set of structurally defined apo-holo pairs with cryptic sites. Next, we comprehensively characterize the cryptic sites in terms of their sequence, structure, and dynamics attributes. We find that cryptic sites tend to be as conserved in evolution as traditional binding pockets but are less hydrophobic and more flexible. Relying on this characterization, we use machine learning to predict cryptic sites with relatively high accuracy (for our benchmark, the true positive and false positive rates are 73% and 29%, respectively). We then predict cryptic sites in the entire structurally characterized human proteome (11,201 structures, covering 23% of all residues in the proteome). CryptoSite increases the size of the potentially "druggable" human proteome from ~40% to ~78% of disease-associated proteins. Finally, to demonstrate the utility of our approach in practice, we experimentally validate a cryptic site in protein tyrosine phosphatase 1B using a covalent ligand and NMR spectroscopy. The CryptoSite Web server is available at http://salilab.org/cryptosite.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
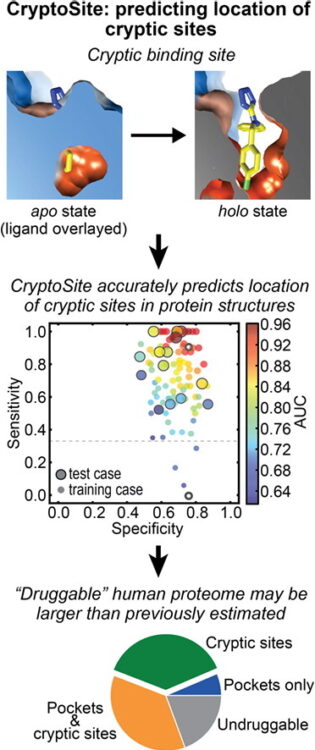
Dominik, Pawel K; Borowska, Marta T; Dalmas, Olivier; Kim, Sangwoo S; Perozo, Eduardo; Keenan, Robert J; Kossiakoff, Anthony A
Conformational Chaperones for Structural Studies of Membrane Proteins Using Antibody Phage Display with Nanodiscs Journal Article
In: Structure, vol. 24, no. 2, pp. 300–309, 2016, ISSN: 1878-4186.
@article{pmid26749445,
title = {Conformational Chaperones for Structural Studies of Membrane Proteins Using Antibody Phage Display with Nanodiscs},
author = {Pawel K Dominik and Marta T Borowska and Olivier Dalmas and Sangwoo S Kim and Eduardo Perozo and Robert J Keenan and Anthony A Kossiakoff},
doi = {10.1016/j.str.2015.11.014},
issn = {1878-4186},
year = {2016},
date = {2016-02-01},
urldate = {2016-02-01},
journal = {Structure},
volume = {24},
number = {2},
pages = {300--309},
abstract = {A major challenge in membrane biophysics is to define the mechanistic linkages between a protein's conformational transitions and its function. We describe a novel approach to stabilize transient functional states of membrane proteins in native-like lipid environments allowing for their structural and biochemical characterization. This is accomplished by combining the power of antibody Fab-based phage display selection with the benefits of embedding membrane protein targets in lipid-filled nanodiscs. In addition to providing a stabilizing lipid environment, nanodiscs afford significant technical advantages over detergent-based formats. This enables the production of a rich pool of high-performance Fab binders that can be used as crystallization chaperones, as fiducial markers for single-particle cryoelectron microscopy, and as probes of different conformational states. Moreover, nanodisc-generated Fabs can be used to identify detergents that best mimic native membrane environments for use in biophysical studies.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
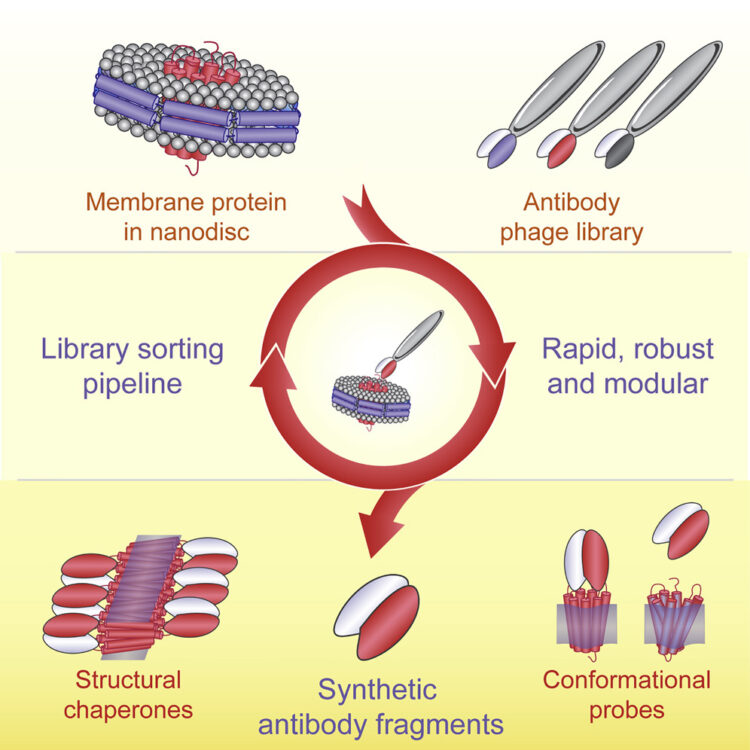
Kuruganti, Srilalitha; Miersch, Shane; Deshpande, Ashlesha; Speir, Jeffrey A; Harris, Bethany D; Schriewer, Jill M; Buller, R Mark L; Sidhu, Sachdev S; Walter, Mark R
Cytokine Activation by Antibody Fragments Targeted to Cytokine-Receptor Signaling Complexes Journal Article
In: J Biol Chem, vol. 291, no. 1, pp. 447–461, 2016, ISSN: 1083-351X.
@article{pmid26546677,
title = {Cytokine Activation by Antibody Fragments Targeted to Cytokine-Receptor Signaling Complexes},
author = {Srilalitha Kuruganti and Shane Miersch and Ashlesha Deshpande and Jeffrey A Speir and Bethany D Harris and Jill M Schriewer and R Mark L Buller and Sachdev S Sidhu and Mark R Walter},
doi = {10.1074/jbc.M115.665943},
issn = {1083-351X},
year = {2016},
date = {2016-01-01},
urldate = {2016-01-01},
journal = {J Biol Chem},
volume = {291},
number = {1},
pages = {447--461},
abstract = {Exogenous cytokine therapy can induce systemic toxicity, which might be prevented by activating endogenously produced cytokines in local cell niches. Here we developed antibody-based activators of cytokine signaling (AcCS), which recognize cytokines only when they are bound to their cell surface receptors. AcCS were developed for type I interferons (IFNs), which induce cellular activities by binding to cell surface receptors IFNAR1 and IFNAR2. As a potential alternative to exogenous IFN therapy, AcCS were shown to potentiate the biological activities of natural IFNs by ∼100-fold. Biochemical and structural characterization demonstrates that the AcCS stabilize the IFN-IFNAR2 binary complex by recognizing an IFN-induced conformational change in IFNAR2. Using IFN mutants that disrupt IFNAR1 binding, AcCS were able to enhance IFN antiviral potency without activating antiproliferative responses. This suggests AcCS can be used to manipulate cytokine signaling for basic science and possibly for therapeutic applications.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
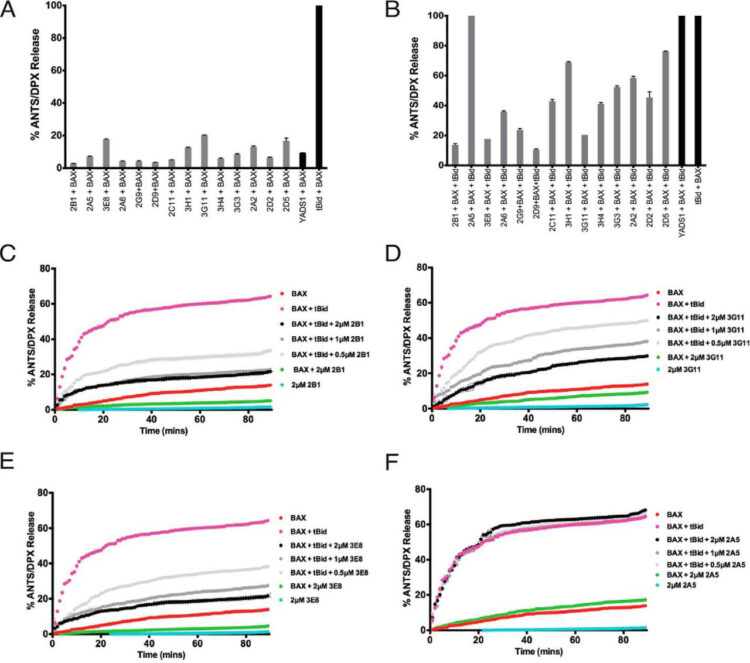
Uchime, Onyinyechukwu; Dai, Zhou; Biris, Nikolaos; Lee, David; Sidhu, Sachdev S; Li, Sheng; Lai, Jonathan R; Gavathiotis, Evripidis
Synthetic Antibodies Inhibit Bcl-2-associated X Protein (BAX) through Blockade of the N-terminal Activation Site Journal Article
In: J Biol Chem, vol. 291, no. 1, pp. 89–102, 2016, ISSN: 1083-351X.
@article{pmid26565029,
title = {Synthetic Antibodies Inhibit Bcl-2-associated X Protein (BAX) through Blockade of the N-terminal Activation Site},
author = {Onyinyechukwu Uchime and Zhou Dai and Nikolaos Biris and David Lee and Sachdev S Sidhu and Sheng Li and Jonathan R Lai and Evripidis Gavathiotis},
doi = {10.1074/jbc.M115.680918},
issn = {1083-351X},
year = {2016},
date = {2016-01-01},
urldate = {2016-01-01},
journal = {J Biol Chem},
volume = {291},
number = {1},
pages = {89--102},
abstract = {The BCL-2 protein family plays a critical role in regulating cellular commitment to mitochondrial apoptosis. Pro-apoptotic Bcl-2-associated X protein (BAX) is an executioner protein of the BCL-2 family that represents the gateway to mitochondrial apoptosis. Following cellular stresses that induce apoptosis, cytosolic BAX is activated and translocates to the mitochondria, where it inserts into the mitochondrial outer membrane to form a toxic pore. How the BAX activation pathway proceeds and how this may be inhibited is not yet completely understood. Here we describe synthetic antibody fragments (Fabs) as structural and biochemical probes to investigate the potential mechanisms of BAX regulation. These synthetic Fabs bind with high affinity to BAX and inhibit its activation by the BH3-only protein tBID (truncated Bcl2 interacting protein) in assays using liposomal membranes. Inhibition of BAX by a representative Fab, 3G11, prevented mitochondrial translocation of BAX and BAX-mediated cytochrome c release. Using NMR and hydrogen-deuterium exchange mass spectrometry, we showed that 3G11 forms a stoichiometric and stable complex without inducing a significant conformational change on monomeric and inactive BAX. We identified that the Fab-binding site on BAX involves residues of helices α1/α6 and the α1-α2 loop. Therefore, the inhibitory binding surface of 3G11 overlaps with the N-terminal activation site of BAX, suggesting a novel mechanism of BAX inhibition through direct binding to the BAX N-terminal activation site. The synthetic Fabs reported here reveal, as probes, novel mechanistic insights into BAX inhibition and provide a blueprint for developing inhibitors of BAX activation.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
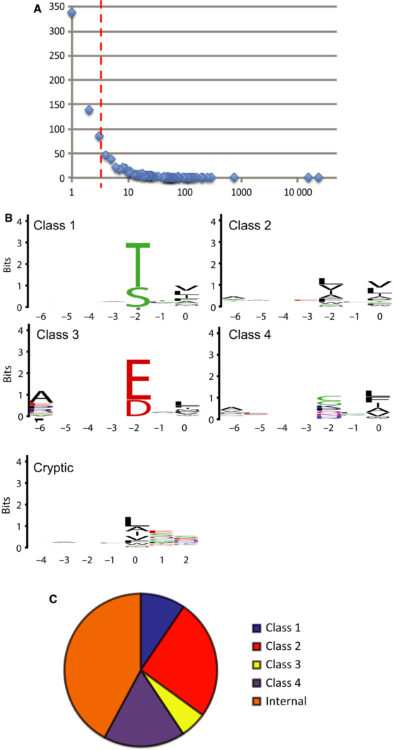
Garrido-Urbani, Sarah; Garg, Pankaj; Ghossoub, Rania; Arnold, Roland; Lembo, Frédérique; Sundell, Gustav N; Kim, Philip M; Lopez, Marc; Zimmermann, Pascale; Sidhu, Sachdev S; Ivarsson, Ylva
Proteomic peptide phage display uncovers novel interactions of the PDZ1-2 supramodule of syntenin Journal Article
In: FEBS Lett, vol. 590, no. 1, pp. 3–12, 2016, ISSN: 1873-3468.
@article{pmid26787460,
title = {Proteomic peptide phage display uncovers novel interactions of the PDZ1-2 supramodule of syntenin},
author = {Sarah Garrido-Urbani and Pankaj Garg and Rania Ghossoub and Roland Arnold and Frédérique Lembo and Gustav N Sundell and Philip M Kim and Marc Lopez and Pascale Zimmermann and Sachdev S Sidhu and Ylva Ivarsson},
doi = {10.1002/1873-3468.12037},
issn = {1873-3468},
year = {2016},
date = {2016-01-01},
urldate = {2016-01-01},
journal = {FEBS Lett},
volume = {590},
number = {1},
pages = {3--12},
abstract = {Syntenin has crucial roles in cell adhesion, cell migration and synaptic transmission. Its closely linked postsynaptic density-95, discs large 1, zonula occludens-1 (PDZ) domains typically interact with C-terminal ligands. We profile syntenin PDZ1-2 through proteomic peptide phage display (ProP-PD) using a library that displays C-terminal regions of the human proteome. The protein recognizes a broad range of peptides, with a preference for hydrophobic motifs and has a tendency to recognize cryptic internal ligands. We validate the interaction with nectin-1 through orthogonal assays. The study demonstrates the power of ProP-PD as a complementary approach to uncover interactions of potential biological relevance.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
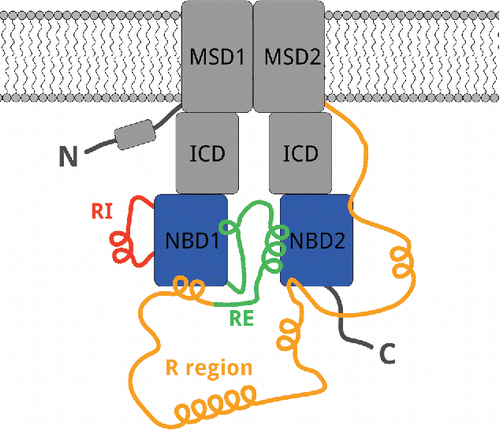
Gakhal, Amandeep K; Jensen, Timothy J; Bozoky, Zoltan; Roldan, Ariel; Lukacs, Gergely L; Forman-Kay, Julie; Riordan, John R; Sidhu, Sachdev S
Development and characterization of synthetic antibodies binding to the cystic fibrosis conductance regulator Journal Article
In: MAbs, vol. 8, no. 6, pp. 1167–1176, 2016, ISSN: 1942-0870.
@article{pmid27185291,
title = {Development and characterization of synthetic antibodies binding to the cystic fibrosis conductance regulator},
author = {Amandeep K Gakhal and Timothy J Jensen and Zoltan Bozoky and Ariel Roldan and Gergely L Lukacs and Julie Forman-Kay and John R Riordan and Sachdev S Sidhu},
doi = {10.1080/19420862.2016.1186320},
issn = {1942-0870},
year = {2016},
date = {2016-01-01},
urldate = {2016-01-01},
journal = {MAbs},
volume = {8},
number = {6},
pages = {1167--1176},
abstract = {Cystic fibrosis transmembrane conductance regulator (CFTR) is a chloride channel in the apical surface of epithelial cells in the airway and gastrointestinal tract, and mutation of CFTR is the underlying cause of cystic fibrosis. However, the precise molecular details of the structure and function of CFTR in native and disease states remains elusive and cystic fibrosis researchers are hindered by a lack of high specificity, high affinity binding reagents for use in structural and biological studies. Here, we describe a panel of synthetic antigen-binding fragments (Fabs) isolated from a phage-displayed library that are specific for intracellular domains of CFTR that include the nucleotide-binding domains (NBD1 and NBD2), the R-region, and the regulatory insertion loop of NBD1. Binding assays performed under conditions that promote the native fold of the protein demonstrated that all Fabs recognized full-length CFTR. However, only the NBD1-specific Fab recognized denatured CFTR by western blot, suggesting a conformational epitope requirement for the other Fabs. Surface plasmon resonance experiments showed that the R-region Fab binds with high affinity to both the phosphorylated and unphosphorylated R-region. In addition, NMR analysis of bound versus unbound R-region revealed a distinct conformational effect upon Fab binding. We further defined residues involved with antibody recognition using an overlapping peptide array. In summary, we describe methodology complementary to previous hybridoma-based efforts to develop antibody reagents to CFTR, and introduce a synthetic antibody panel to aid structural and biological studies.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Nilvebrant, Johan; Tessier, Peter M; Sidhu, Sachdev S
Engineered Autonomous Human Variable Domains Journal Article
In: Curr Pharm Des, vol. 22, no. 43, pp. 6527–6537, 2016, ISSN: 1873-4286.
@article{pmid27655414,
title = {Engineered Autonomous Human Variable Domains},
author = {Johan Nilvebrant and Peter M Tessier and Sachdev S Sidhu},
doi = {10.2174/1381612822666160921143011},
issn = {1873-4286},
year = {2016},
date = {2016-01-01},
journal = {Curr Pharm Des},
volume = {22},
number = {43},
pages = {6527--6537},
abstract = {BACKGROUND: The complex multi-chain architecture of antibodies has spurred interest in smaller derivatives that retain specificity but can be more easily produced in bacteria. Domain antibodies consisting of single variable domains are the smallest antibody fragments and have been shown to possess enhanced ability to target epitopes that are difficult to access using multidomain antibodies. However, in contrast to natural camelid antibody domains, human variable domains typically suffer from low stability and high propensity to aggregate.nnMETHODS: This review summarizes strategies to improve the biophysical properties of heavy chain variable domains from human antibodies with an emphasis on aggregation resistance. Several protein engineering approaches have targeted antibody frameworks and complementarity determining regions to stabilize the native state and prevent aggregation of the denatured state.nnCONCLUSION: Recent findings enable the construction of highly diverse libraries enriched in aggregation-resistant variants that are expected to provide binders to diverse antigens. Engineered domain antibodies possess unique advantages in expression, epitope preference and flexibility of formatting over conventional immunoreagents and are a promising class of antibody fragments for biomedical development.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}