Publications: 2013
2013
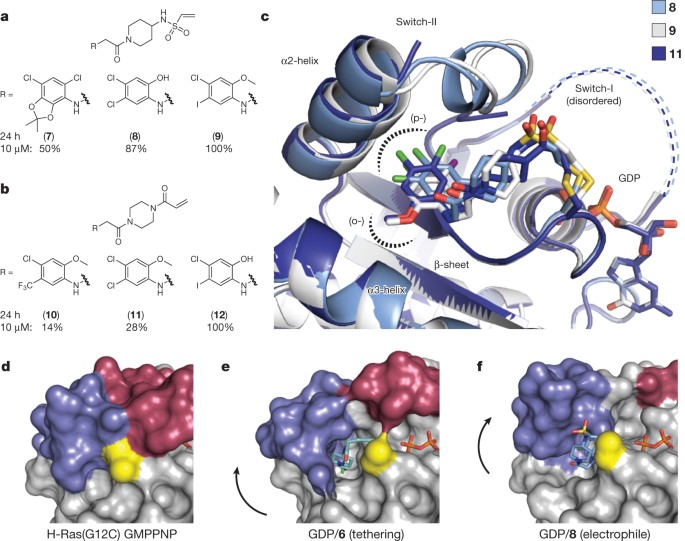
Ostrem, Jonathan M; Peters, Ulf; Sos, Martin L; Wells, James A; Shokat, Kevan M
K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions Journal Article
In: Nature, vol. 503, no. 7477, pp. 548–551, 2013, ISSN: 1476-4687.
@article{pmid24256730,
title = {K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions},
author = {Jonathan M Ostrem and Ulf Peters and Martin L Sos and James A Wells and Kevan M Shokat},
doi = {10.1038/nature12796},
issn = {1476-4687},
year = {2013},
date = {2013-11-01},
urldate = {2013-11-01},
journal = {Nature},
volume = {503},
number = {7477},
pages = {548--551},
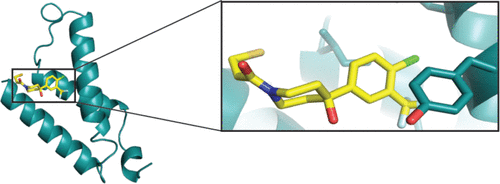
abstract = {Somatic mutations in the small GTPase K-Ras are the most common activating lesions found in human cancer, and are generally associated with poor response to standard therapies. Efforts to target this oncogene directly have faced difficulties owing to its picomolar affinity for GTP/GDP and the absence of known allosteric regulatory sites. Oncogenic mutations result in functional activation of Ras family proteins by impairing GTP hydrolysis. With diminished regulation by GTPase activity, the nucleotide state of Ras becomes more dependent on relative nucleotide affinity and concentration. This gives GTP an advantage over GDP and increases the proportion of active GTP-bound Ras. Here we report the development of small molecules that irreversibly bind to a common oncogenic mutant, K-Ras(G12C). These compounds rely on the mutant cysteine for binding and therefore do not affect the wild-type protein. Crystallographic studies reveal the formation of a new pocket that is not apparent in previous structures of Ras, beneath the effector binding switch-II region. Binding of these inhibitors to K-Ras(G12C) disrupts both switch-I and switch-II, subverting the native nucleotide preference to favour GDP over GTP and impairing binding to Raf. Our data provide structure-based validation of a new allosteric regulatory site on Ras that is targetable in a mutant-specific manner.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Wiita, Arun P; Ziv, Etay; Wiita, Paul J; Urisman, Anatoly; Julien, Olivier; Burlingame, Alma L; Weissman, Jonathan S; Wells, James A
Global cellular response to chemotherapy-induced apoptosis Journal Article
In: Elife, vol. 2, pp. e01236, 2013, ISSN: 2050-084X.
@article{pmid24171104,
title = {Global cellular response to chemotherapy-induced apoptosis},
author = {Arun P Wiita and Etay Ziv and Paul J Wiita and Anatoly Urisman and Olivier Julien and Alma L Burlingame and Jonathan S Weissman and James A Wells},
doi = {10.7554/eLife.01236},
issn = {2050-084X},
year = {2013},
date = {2013-10-01},
urldate = {2013-10-01},
journal = {Elife},
volume = {2},
pages = {e01236},
abstract = {How cancer cells globally struggle with a chemotherapeutic insult before succumbing to apoptosis is largely unknown. Here we use an integrated systems-level examination of transcription, translation, and proteolysis to understand these events central to cancer treatment. As a model we study myeloma cells exposed to the proteasome inhibitor bortezomib, a first-line therapy. Despite robust transcriptional changes, unbiased quantitative proteomics detects production of only a few critical anti-apoptotic proteins against a background of general translation inhibition. Simultaneous ribosome profiling further reveals potential translational regulation of stress response genes. Once the apoptotic machinery is engaged, degradation by caspases is largely independent of upstream bortezomib effects. Moreover, previously uncharacterized non-caspase proteolytic events also participate in cellular deconstruction. Our systems-level data also support co-targeting the anti-apoptotic regulator HSF1 to promote cell death by bortezomib. This integrated approach offers unique, in-depth insight into apoptotic dynamics that may prove important to preclinical evaluation of any anti-cancer compound. DOI:http://dx.doi.org/10.7554/eLife.01236.001.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

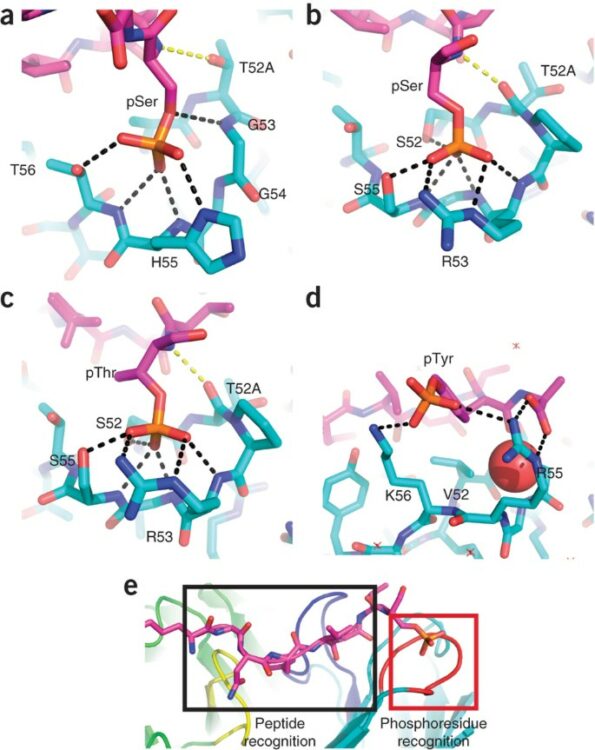
Koerber, James T; Thomsen, Nathan D; Hannigan, Brett T; Degrado, William F; Wells, James A
Nature-inspired design of motif-specific antibody scaffolds Journal Article
In: Nat Biotechnol, vol. 31, no. 10, pp. 916–921, 2013, ISSN: 1546-1696.
@article{pmid23955275,
title = {Nature-inspired design of motif-specific antibody scaffolds},
author = {James T Koerber and Nathan D Thomsen and Brett T Hannigan and William F Degrado and James A Wells},
doi = {10.1038/nbt.2672},
issn = {1546-1696},
year = {2013},
date = {2013-10-01},
urldate = {2013-10-01},
journal = {Nat Biotechnol},
volume = {31},
number = {10},
pages = {916--921},
abstract = {Aberrant changes in post-translational modifications (PTMs) such as phosphate groups underlie a majority of human diseases. However, detection and quantification of PTMs for diagnostic or biomarker applications often require PTM-specific monoclonal antibodies (mAbs), which are challenging to generate using traditional antibody-selection methods. Here we outline a general strategy for producing synthetic, PTM-specific mAbs by engineering a motif-specific 'hot spot' into an antibody scaffold. Inspired by a natural phosphate-binding motif, we designed and selected mAb scaffolds with hot spots specific for phosphoserine, phosphothreonine or phosphotyrosine. Crystal structures of the phospho-specific mAbs revealed two distinct modes of phosphoresidue recognition. Our data suggest that each hot spot functions independently of the surrounding scaffold, as phage display antibody libraries using these scaffolds yielded >50 phospho- and target-specific mAbs against 70% of target peptides. Our motif-specific scaffold strategy may provide a general solution for rapid, robust development of anti-PTM mAbs for signaling, diagnostic and therapeutic applications.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Carter, Paul J; Hazuda, Daria; Wells, James A
Next generation therapeutics Miscellaneous
2013, ISSN: 1879-0402.
@misc{pmid23683350,
title = {Next generation therapeutics},
author = {Paul J Carter and Daria Hazuda and James A Wells},
doi = {10.1016/j.cbpa.2013.04.022},
issn = {1879-0402},
year = {2013},
date = {2013-06-01},
journal = {Curr Opin Chem Biol},
volume = {17},
number = {3},
pages = {317--319},
keywords = {},
pubstate = {published},
tppubtype = {misc}
}
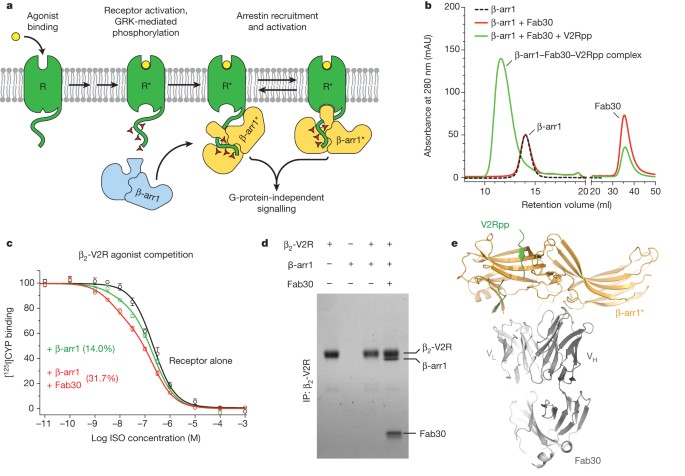
Shukla, Arun K; Manglik, Aashish; Kruse, Andrew C; Xiao, Kunhong; Reis, Rosana I; Tseng, Wei-Chou; Staus, Dean P; Hilger, Daniel; Uysal, Serdar; Huang, Li-Yin; Paduch, Marcin; Tripathi-Shukla, Prachi; Koide, Akiko; Koide, Shohei; Weis, William I; Kossiakoff, Anthony A; Kobilka, Brian K; Lefkowitz, Robert J
Structure of active β-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide Journal Article
In: Nature, vol. 497, no. 7447, pp. 137–141, 2013, ISSN: 1476-4687.
@article{pmid23604254,
title = {Structure of active β-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide},
author = {Arun K Shukla and Aashish Manglik and Andrew C Kruse and Kunhong Xiao and Rosana I Reis and Wei-Chou Tseng and Dean P Staus and Daniel Hilger and Serdar Uysal and Li-Yin Huang and Marcin Paduch and Prachi Tripathi-Shukla and Akiko Koide and Shohei Koide and William I Weis and Anthony A Kossiakoff and Brian K Kobilka and Robert J Lefkowitz},
doi = {10.1038/nature12120},
issn = {1476-4687},
year = {2013},
date = {2013-05-01},
urldate = {2013-05-01},
journal = {Nature},
volume = {497},
number = {7447},
pages = {137--141},
abstract = {The functions of G-protein-coupled receptors (GPCRs) are primarily mediated and modulated by three families of proteins: the heterotrimeric G proteins, the G-protein-coupled receptor kinases (GRKs) and the arrestins. G proteins mediate activation of second-messenger-generating enzymes and other effectors, GRKs phosphorylate activated receptors, and arrestins subsequently bind phosphorylated receptors and cause receptor desensitization. Arrestins activated by interaction with phosphorylated receptors can also mediate G-protein-independent signalling by serving as adaptors to link receptors to numerous signalling pathways. Despite their central role in regulation and signalling of GPCRs, a structural understanding of β-arrestin activation and interaction with GPCRs is still lacking. Here we report the crystal structure of β-arrestin-1 (also called arrestin-2) in complex with a fully phosphorylated 29-amino-acid carboxy-terminal peptide derived from the human V2 vasopressin receptor (V2Rpp). This peptide has previously been shown to functionally and conformationally activate β-arrestin-1 (ref. 5). To capture this active conformation, we used a conformationally selective synthetic antibody fragment (Fab30) that recognizes the phosphopeptide-activated state of β-arrestin-1. The structure of the β-arrestin-1-V2Rpp-Fab30 complex shows marked conformational differences in β-arrestin-1 compared to its inactive conformation. These include rotation of the amino- and carboxy-terminal domains relative to each other, and a major reorientation of the 'lariat loop' implicated in maintaining the inactive state of β-arrestin-1. These results reveal, at high resolution, a receptor-interacting interface on β-arrestin, and they indicate a potentially general molecular mechanism for activation of these multifunctional signalling and regulatory proteins.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Yang, Cindy F; Chiang, Michael C; Gray, Daniel C; Prabhakaran, Mahalakshmi; Alvarado, Maricruz; Juntti, Scott A; Unger, Elizabeth K; Wells, James A; Shah, Nirao M
Sexually dimorphic neurons in the ventromedial hypothalamus govern mating in both sexes and aggression in males Journal Article
In: Cell, vol. 153, no. 4, pp. 896–909, 2013, ISSN: 1097-4172.
@article{pmid23663785,
title = {Sexually dimorphic neurons in the ventromedial hypothalamus govern mating in both sexes and aggression in males},
author = {Cindy F Yang and Michael C Chiang and Daniel C Gray and Mahalakshmi Prabhakaran and Maricruz Alvarado and Scott A Juntti and Elizabeth K Unger and James A Wells and Nirao M Shah},
doi = {10.1016/j.cell.2013.04.017},
issn = {1097-4172},
year = {2013},
date = {2013-05-01},
urldate = {2013-05-01},
journal = {Cell},
volume = {153},
number = {4},
pages = {896--909},
abstract = {Sexual dimorphisms in the brain underlie behavioral sex differences, but the function of individual sexually dimorphic neuronal populations is poorly understood. Neuronal sexual dimorphisms typically represent quantitative differences in cell number, gene expression, or other features, and it is unknown whether these dimorphisms control sex-typical behavior exclusively in one sex or in both sexes. The progesterone receptor (PR) controls female sexual behavior, and we find many sex differences in number, distribution, or projections of PR-expressing neurons in the adult mouse brain. Using a genetic strategy we developed, we have ablated one such dimorphic PR-expressing neuronal population located in the ventromedial hypothalamus (VMH). Ablation of these neurons in females greatly diminishes sexual receptivity. Strikingly, the corresponding ablation in males reduces mating and aggression. Our findings reveal the functions of a molecularly defined, sexually dimorphic neuronal population in the brain. Moreover, we show that sexually dimorphic neurons can control distinct sex-typical behaviors in both sexes.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
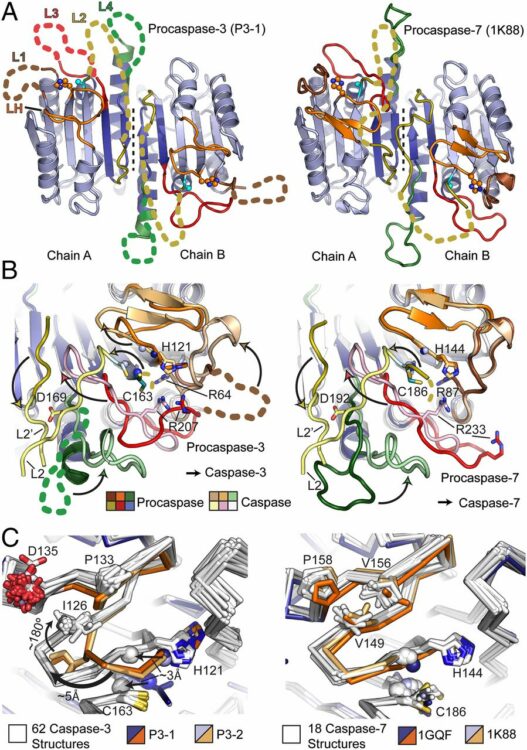
Thomsen, Nathan D; Koerber, James T; Wells, James A
Structural snapshots reveal distinct mechanisms of procaspase-3 and -7 activation Journal Article
In: Proc Natl Acad Sci U S A, vol. 110, no. 21, pp. 8477–8482, 2013, ISSN: 1091-6490.
@article{pmid23650375,
title = {Structural snapshots reveal distinct mechanisms of procaspase-3 and -7 activation},
author = {Nathan D Thomsen and James T Koerber and James A Wells},
doi = {10.1073/pnas.1306759110},
issn = {1091-6490},
year = {2013},
date = {2013-05-01},
urldate = {2013-05-01},
journal = {Proc Natl Acad Sci U S A},
volume = {110},
number = {21},
pages = {8477--8482},
abstract = {Procaspase-3 (P3) and procaspase-7 (P7) are activated through proteolytic maturation to form caspase-3 (C3) and caspase-7 (C7), respectively, which serve overlapping but nonredundant roles as the executioners of apoptosis in humans. However, it is unclear if differences in P3 and P7 maturation mechanisms underlie their unique biological functions, as the structure of P3 remains unknown. Here, we report structures of P3 in a catalytically inactive conformation, structures of P3 and P7 bound to covalent peptide inhibitors that reveal the active conformation of the zymogens, and the structure of a partially matured C7:P7 heterodimer. Along with a biochemical analysis, we show that P3 is catalytically inactive and matures through a symmetric all-or-nothing process. In contrast, P7 contains latent catalytic activity and matures through an asymmetric and tiered mechanism, suggesting a lower threshold for activation. Finally, we use our structures to design a selection strategy for conformation specific antibody fragments that stimulate procaspase activity, showing that executioner procaspase conformational equilibrium can be rationally modulated. Our studies provide a structural framework that may help to explain the unique roles of these important proapoptotic enzymes, and suggest general strategies for the discovery of proenzyme activators.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
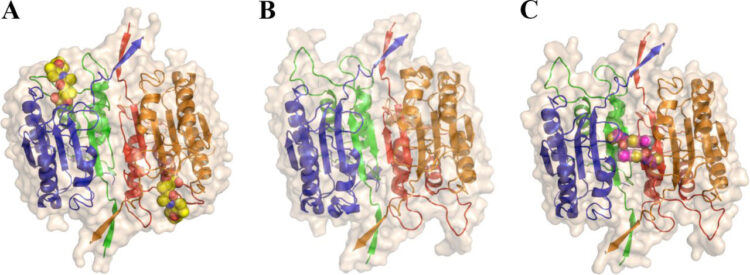
Datta, Debajyoti; McClendon, Christopher L; Jacobson, Matthew P; Wells, James A
Substrate and inhibitor-induced dimerization and cooperativity in caspase-1 but not caspase-3 Journal Article
In: J Biol Chem, vol. 288, no. 14, pp. 9971–9981, 2013, ISSN: 1083-351X.
@article{pmid23386603,
title = {Substrate and inhibitor-induced dimerization and cooperativity in caspase-1 but not caspase-3},
author = {Debajyoti Datta and Christopher L McClendon and Matthew P Jacobson and James A Wells},
doi = {10.1074/jbc.M112.426460},
issn = {1083-351X},
year = {2013},
date = {2013-04-01},
urldate = {2013-04-01},
journal = {J Biol Chem},
volume = {288},
number = {14},
pages = {9971--9981},
abstract = {Caspases are intracellular cysteine-class proteases with aspartate specificity that is critical for driving processes as diverse as the innate immune response and apoptosis, exemplified by caspase-1 and caspase-3, respectively. Interestingly, caspase-1 cleaves far fewer cellular substrates than caspase-3 and also shows strong positive cooperativity between the two active sites of the homodimer, unlike caspase-3. Biophysical and kinetic studies here present a molecular basis for this difference. Analytical ultracentrifugation experiments show that mature caspase-1 exists predominantly as a monomer under physiological concentrations that undergoes dimerization in the presence of substrate; specifically, substrate binding shifts the KD for dimerization by 20-fold. We have created a hemi-active site-labeled dimer of caspase-1, where one site is blocked with the covalent active site inhibitor, benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone. This hemi-labeled enzyme is about 9-fold more active than the apo-dimer of caspase-1. These studies suggest that substrate not only drives dimerization but also, once bound to one site in the dimer, promotes an active conformation in the other monomer. Steady-state kinetic analysis and modeling independently support this model, where binding of one substrate molecule not only increases substrate binding in preformed dimers but also drives the formation of heterodimers. Thus, the cooperativity in caspase-1 is driven both by substrate-induced dimerization as well as substrate-induced activation. Substrate-induced dimerization and activation seen in caspase-1 and not in caspase-3 may reflect their biological roles. Whereas caspase-1 cleaves a dramatically smaller number of cellular substrates that need to be concentrated near inflammasomes, caspase-3 is a constitutively active dimer that cleaves many more substrates located diffusely throughout the cell.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Paduch, Marcin; Koide, Akiko; Uysal, Serdar; Rizk, Shahir S; Koide, Shohei; Kossiakoff, Anthony A
Generating conformation-specific synthetic antibodies to trap proteins in selected functional states Journal Article
In: Methods, vol. 60, no. 1, pp. 3–14, 2013, ISSN: 1095-9130.
@article{pmid23280336,
title = {Generating conformation-specific synthetic antibodies to trap proteins in selected functional states},
author = {Marcin Paduch and Akiko Koide and Serdar Uysal and Shahir S Rizk and Shohei Koide and Anthony A Kossiakoff},
doi = {10.1016/j.ymeth.2012.12.010},
issn = {1095-9130},
year = {2013},
date = {2013-03-01},
urldate = {2013-03-01},
journal = {Methods},
volume = {60},
number = {1},
pages = {3--14},
abstract = {A set of phage display sorting strategies and validation methodologies are presented that are capable of producing high performance synthetic antibodies (sABs) with customized properties. Exquisite control of antigen and conditions during the phage display selection process can yield sABs that: (1) recognize conformational states, (2) target specific regions of the surface of a protein, (3) induce conformational changes, and (4) capture and stabilize multi-protein complexes. These unique capabilities open myriad opportunities to study complex macromolecular processes inaccessible to traditional affinity reagent technology. We present detailed protocols for de novo isolation of binders, as well as examples of downstream biophysical characterization. The methods described are generalizable and can be adapted to other in vitro direct evolution approaches based on yeast or mRNA display.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Wang, Ningkun; Majmudar, Chinmay Y; Pomerantz, William C; Gagnon, Jessica K; Sadowsky, Jack D; Meagher, Jennifer L; Johnson, Taylor K; Stuckey, Jeanne A; Brooks, Charles L; Wells, James A; Mapp, Anna K
Ordering a dynamic protein via a small-molecule stabilizer Journal Article
In: J Am Chem Soc, vol. 135, no. 9, pp. 3363–3366, 2013, ISSN: 1520-5126.
@article{pmid23384013,
title = {Ordering a dynamic protein via a small-molecule stabilizer},
author = {Ningkun Wang and Chinmay Y Majmudar and William C Pomerantz and Jessica K Gagnon and Jack D Sadowsky and Jennifer L Meagher and Taylor K Johnson and Jeanne A Stuckey and Charles L Brooks and James A Wells and Anna K Mapp},
doi = {10.1021/ja3122334},
issn = {1520-5126},
year = {2013},
date = {2013-03-01},
urldate = {2013-03-01},
journal = {J Am Chem Soc},
volume = {135},
number = {9},
pages = {3363--3366},
abstract = {Like many coactivators, the GACKIX domain of the master coactivator CBP/p300 recognizes transcriptional activators of diverse sequence composition via dynamic binding surfaces. The conformational dynamics of GACKIX that underlie its function also render it especially challenging for structural characterization. We have found that the ligand discovery strategy of Tethering is an effective method for identifying small-molecule fragments that stabilize the GACKIX domain, enabling for the first time the crystallographic characterization of this important motif. The 2.0 Å resolution structure of GACKIX complexed to a small molecule was further analyzed by molecular dynamics simulations, which revealed the importance of specific side-chain motions that remodel the activator binding site in order to accommodate binding partners of distinct sequence and size. More broadly, these results suggest that Tethering can be a powerful strategy for identifying small-molecule stabilizers of conformationally malleable proteins, thus facilitating their structural characterization and accelerating the discovery of small-molecule modulators.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Crawford, Emily D; Seaman, Julia E; Agard, Nick; Hsu, Gerald W; Julien, Olivier; Mahrus, Sami; Nguyen, Huy; Shimbo, Kazutaka; Yoshihara, Hikari A I; Zhuang, Min; Chalkley, Robert J; Wells, James A
The DegraBase: a database of proteolysis in healthy and apoptotic human cells Journal Article
In: Mol Cell Proteomics, vol. 12, no. 3, pp. 813–824, 2013, ISSN: 1535-9484.
@article{pmid23264352,
title = {The DegraBase: a database of proteolysis in healthy and apoptotic human cells},
author = {Emily D Crawford and Julia E Seaman and Nick Agard and Gerald W Hsu and Olivier Julien and Sami Mahrus and Huy Nguyen and Kazutaka Shimbo and Hikari A I Yoshihara and Min Zhuang and Robert J Chalkley and James A Wells},
doi = {10.1074/mcp.O112.024372},
issn = {1535-9484},
year = {2013},
date = {2013-03-01},
urldate = {2013-03-01},
journal = {Mol Cell Proteomics},
volume = {12},
number = {3},
pages = {813--824},
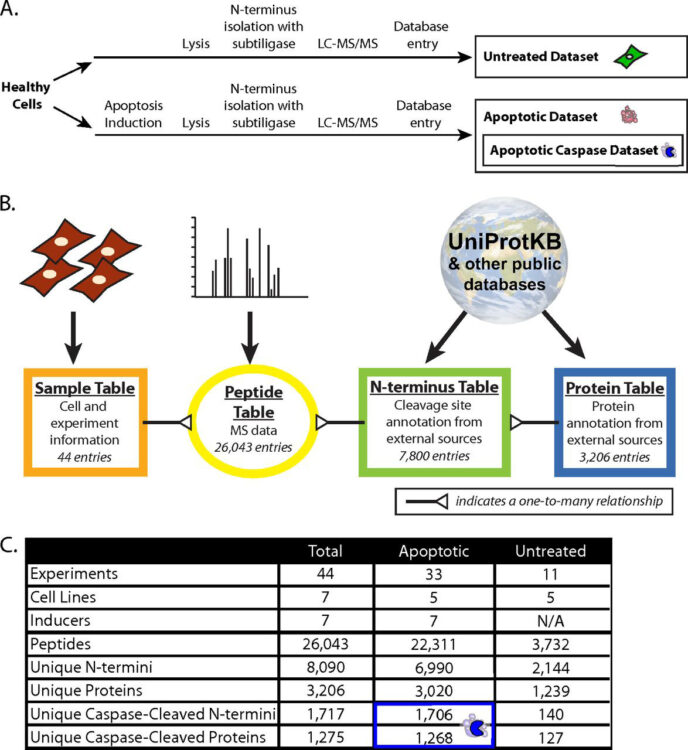
abstract = {Proteolysis is a critical post-translational modification for regulation of cellular processes. Our lab has previously developed a technique for specifically labeling unmodified protein N termini, the α-aminome, using the engineered enzyme, subtiligase. Here we present a database, called the DegraBase (http://wellslab.ucsf.edu/degrabase/), which compiles 8090 unique N termini from 3206 proteins directly identified in subtiligase-based positive enrichment mass spectrometry experiments in healthy and apoptotic human cell lines. We include both previously published and unpublished data in our analysis, resulting in a total of 2144 unique α-amines identified in healthy cells, and 6990 in cells undergoing apoptosis. The N termini derive from three general categories of proteolysis with respect to cleavage location and functional role: translational N-terminal methionine processing (∼10% of total proteolysis), sites close to the translational N terminus that likely represent removal of transit or signal peptides (∼25% of total), and finally, other endoproteolytic cuts (∼65% of total). Induction of apoptosis causes relatively little change in the first two proteolytic categories, but dramatic changes are seen in endoproteolysis. For example, we observed 1706 putative apoptotic caspase cuts, more than double the total annotated sites in the CASBAH and MEROPS databases. In the endoproteolysis category, there are a total of nearly 3000 noncaspase nontryptic cleavages that are not currently reported in the MEROPS database. These studies significantly increase the annotation for all categories of proteolysis in human cells and allow public access for investigators to explore interesting proteolytic events in healthy and apoptotic human cells.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Persson, Helena; Ye, Wei; Wernimont, Amy; Adams, Jarrett J; Koide, Akiko; Koide, Shohei; Lam, Robert; Sidhu, Sachdev S
CDR-H3 diversity is not required for antigen recognition by synthetic antibodies Journal Article
In: J Mol Biol, vol. 425, no. 4, pp. 803–811, 2013, ISSN: 1089-8638.
@article{pmid23219464,
title = {CDR-H3 diversity is not required for antigen recognition by synthetic antibodies},
author = {Helena Persson and Wei Ye and Amy Wernimont and Jarrett J Adams and Akiko Koide and Shohei Koide and Robert Lam and Sachdev S Sidhu},
doi = {10.1016/j.jmb.2012.11.037},
issn = {1089-8638},
year = {2013},
date = {2013-02-01},
urldate = {2013-02-01},
journal = {J Mol Biol},
volume = {425},
number = {4},
pages = {803--811},
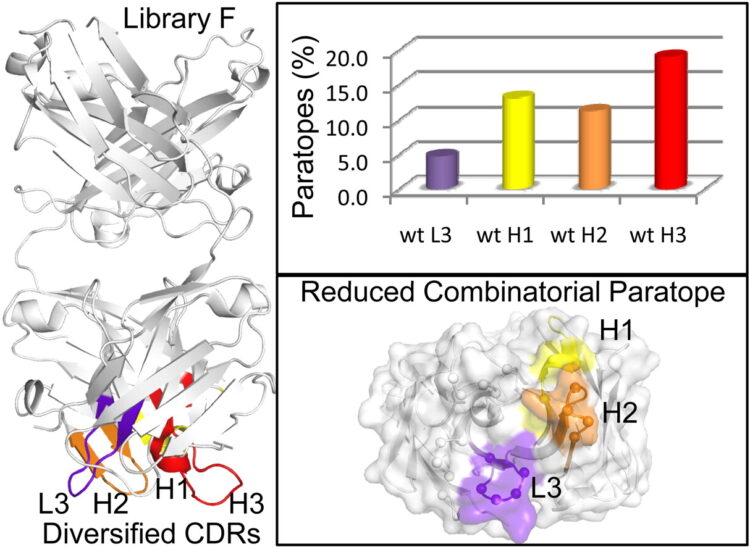
abstract = {A synthetic phage-displayed antibody repertoire was constructed with equivalent chemical diversity in the third complementarity-determining regions of the heavy (CDR-H3) and light (CDR-L3) chains, which contrasts with natural antibodies in which CDR-H3 is much more diverse than CDR-L3 due to the genetic mechanisms that generate antibody encoding genes. Surprisingly, the synthetic repertoire yielded numerous functional antibodies that contained mutated CDR-L3 sequences but a fixed CDR-H3 sequence. Alanine-scanning analysis of antibodies that recognized 10 different antigens but contained a common CDR-H3 loop showed that, in most cases, the fixed CDR-H3 sequence was able to contribute favorably to antigen recognition, but in some cases, the loop was functionally inert. Structural analysis of one such antibody in complex with antigen showed that the inert CDR-H3 loop was nonetheless highly buried at the antibody-antigen interface. Taken together, these results show that CDR-H3 diversity is not necessarily required for the generation of antibodies that recognize diverse protein antigens with high affinity and specificity, and if given the chance, CDR-L3 readily assumes the dominant role for antigen recognition. These results contrast with the commonly accepted view of antigen recognition derived from the analysis of natural antibodies, in which CDR-H3 is presumed to be dominant and CDR-L3 is presumed to play an auxiliary role. Furthermore, the results show that natural antibody function is genetically constrained, and it should be possible to develop more functional synthetic antibody libraries by expanding the diversity of CDR-L3 beyond what is observed in nature.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Zhuang, Min; Guan, Shenheng; Wang, Haopeng; Burlingame, Alma L; Wells, James A
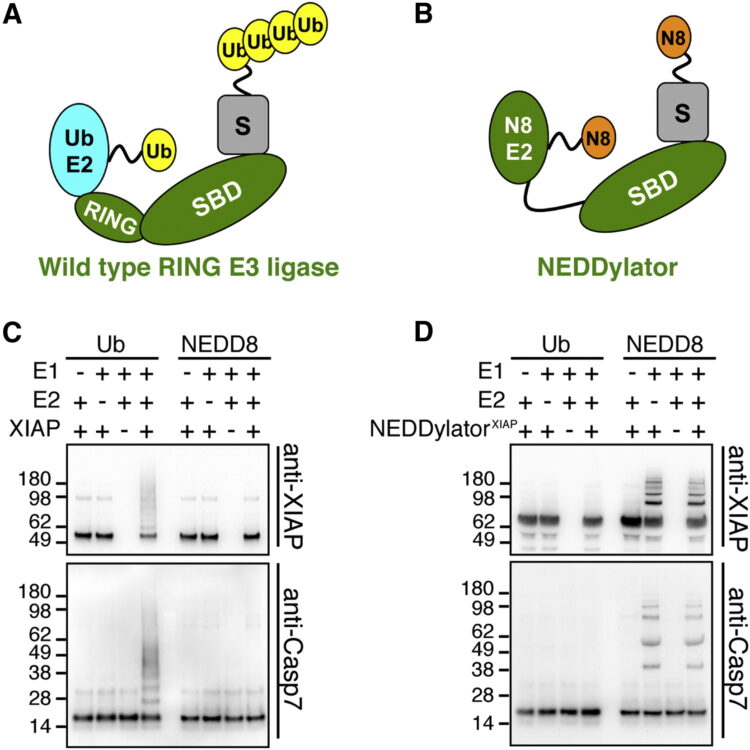
Substrates of IAP ubiquitin ligases identified with a designed orthogonal E3 ligase, the NEDDylator Journal Article
In: Mol Cell, vol. 49, no. 2, pp. 273–282, 2013, ISSN: 1097-4164.
@article{pmid23201124,
title = {Substrates of IAP ubiquitin ligases identified with a designed orthogonal E3 ligase, the NEDDylator},
author = {Min Zhuang and Shenheng Guan and Haopeng Wang and Alma L Burlingame and James A Wells},
doi = {10.1016/j.molcel.2012.10.022},
issn = {1097-4164},
year = {2013},
date = {2013-01-01},
urldate = {2013-01-01},
journal = {Mol Cell},
volume = {49},
number = {2},
pages = {273--282},
abstract = {Inhibitors of Apoptosis Protein (IAPs) are guardian ubiquitin ligases that keep classic proapoptotic proteins in check. Systematic identification of additional IAP substrates is challenged by the heterogeneity and sheer number of ubiquitinated proteins (>5,000). Here we report a powerful catalytic tagging tool, the NEDDylator, which fuses a NEDD8 E2-conjugating enzyme, Ubc12, to the ubiquitin ligase, XIAP or cIAP1. This permits transfer of the rare ubiquitin homolog NEDD8 to the ubiquitin E3 substrates, allowing them to be efficiently purified for LC-MS/MS identification. We have identified >50 potential IAP substrates of both cytosolic and mitochondrial origin that bear hallmark N-terminal IAP binding motifs. These substrates include the recently discovered protein phosphatase PGAM5, which we show is proteolytically processed, accumulates in cytosol during apoptosis, and sensitizes cells to death. These studies reveal mechanisms and antagonistic partners for specific IAPs, and provide a powerful technology for labeling binding partners in transient protein-protein complexes.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}