Publications
View by year
- Publications: 2025
- Publications: 2024
- Publications: 2023
- Publications: 2022
- Publications: 2021
- Publications: 2020
- Publications: 2019
- Publications: 2018
- Publications: 2017
- Publications: 2016
- Publications: 2015
- Publications: 2014
- Publications: 2013
- Publications: 2012
Search
Search for author, keywords, or any other term.
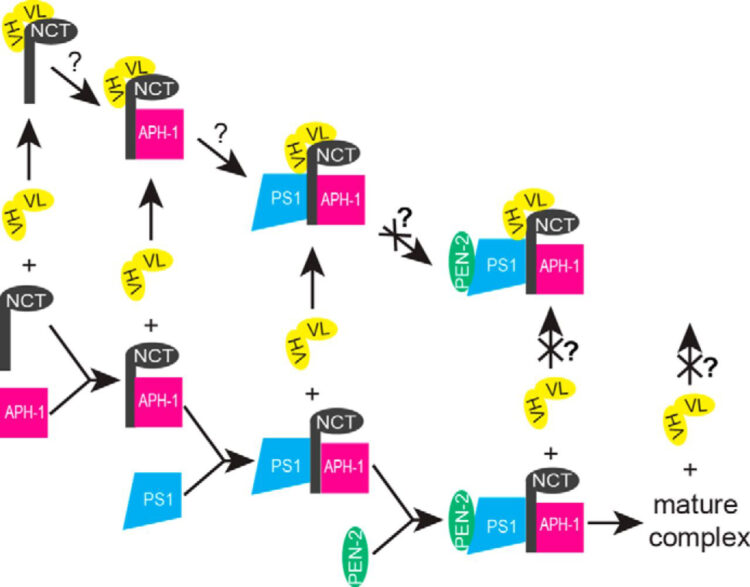
Zhang, Xulun; Hoey, Robert; Koide, Akiko; Dolios, Georgia; Paduch, Marcin; Nguyen, Phuong; Wu, Xianzhong; Li, Yueming; Wagner, Steven L; Wang, Rong; Koide, Shohei; Sisodia, Sangram S
A synthetic antibody fragment targeting nicastrin affects assembly and trafficking of γ-secretase Journal Article
In: J Biol Chem, vol. 289, no. 50, pp. 34851–34861, 2014, ISSN: 1083-351X.
@article{pmid25352592,
title = {A synthetic antibody fragment targeting nicastrin affects assembly and trafficking of γ-secretase},
author = {Xulun Zhang and Robert Hoey and Akiko Koide and Georgia Dolios and Marcin Paduch and Phuong Nguyen and Xianzhong Wu and Yueming Li and Steven L Wagner and Rong Wang and Shohei Koide and Sangram S Sisodia},
doi = {10.1074/jbc.M114.609636},
issn = {1083-351X},
year = {2014},
date = {2014-12-01},
urldate = {2014-12-01},
journal = {J Biol Chem},
volume = {289},
number = {50},
pages = {34851--34861},
abstract = {The γ-secretase complex, composed of presenilin, nicastrin (NCT), anterior pharynx-defective 1 (APH-1), and presenilin enhancer 2 (PEN-2), is assembled in a highly regulated manner and catalyzes the intramembranous proteolysis of many type I membrane proteins, including Notch and amyloid precursor protein. The Notch family of receptors plays important roles in cell fate specification during development and in adult tissues, and aberrant hyperactive Notch signaling causes some forms of cancer. γ-Secretase-mediated processing of Notch at the cell surface results in the generation of the Notch intracellular domain, which associates with several transcriptional coactivators involved in nuclear signaling events. On the other hand, γ-secretase-mediated processing of amyloid precursor protein leads to the production of amyloid β (Aβ) peptides that play an important role in the pathogenesis of Alzheimer disease. We used a phage display approach to identify synthetic antibodies that specifically target NCT and expressed them in the single-chain variable fragment (scFv) format in mammalian cells. We show that expression of a NCT-specific scFv clone, G9, in HEK293 cells decreased the production of the Notch intracellular domain but not the production of amyloid β peptides that occurs in endosomal and recycling compartments. Biochemical studies revealed that scFvG9 impairs the maturation of NCT by associating with immature forms of NCT and, consequently, prevents its association with the other components of the γ-secretase complex, leading to degradation of these molecules. The reduced cell surface levels of mature γ-secretase complexes, in turn, compromise the intramembranous processing of Notch.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
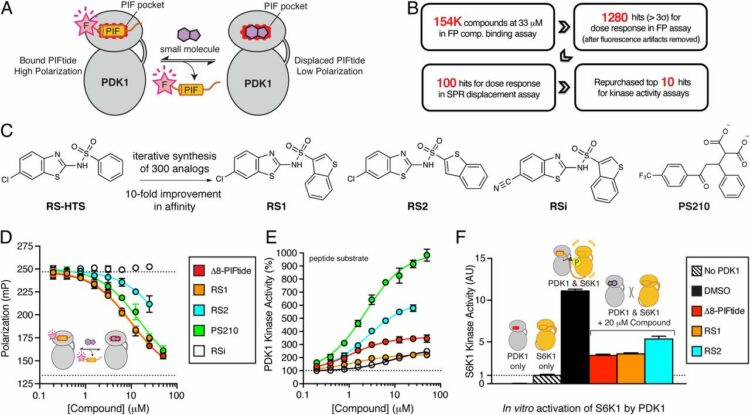
Rettenmaier, T Justin; Sadowsky, Jack D; Thomsen, Nathan D; Chen, Steven C; Doak, Allison K; Arkin, Michelle R; Wells, James A
A small-molecule mimic of a peptide docking motif inhibits the protein kinase PDK1 Journal Article
In: Proc Natl Acad Sci U S A, vol. 111, no. 52, pp. 18590–18595, 2014, ISSN: 1091-6490.
@article{pmid25518860,
title = {A small-molecule mimic of a peptide docking motif inhibits the protein kinase PDK1},
author = {T Justin Rettenmaier and Jack D Sadowsky and Nathan D Thomsen and Steven C Chen and Allison K Doak and Michelle R Arkin and James A Wells},
doi = {10.1073/pnas.1415365112},
issn = {1091-6490},
year = {2014},
date = {2014-12-01},
urldate = {2014-12-01},
journal = {Proc Natl Acad Sci U S A},
volume = {111},
number = {52},
pages = {18590--18595},
abstract = {There is great interest in developing selective protein kinase inhibitors by targeting allosteric sites, but these sites often involve protein-protein or protein-peptide interfaces that are very challenging to target with small molecules. Here we present a systematic approach to targeting a functionally conserved allosteric site on the protein kinase PDK1 called the PDK1-interacting fragment (PIF)tide-binding site, or PIF pocket. More than two dozen prosurvival and progrowth kinases dock a conserved peptide tail into this binding site, which recruits them to PDK1 to become activated. Using a site-directed chemical screen, we identified and chemically optimized ligand-efficient, selective, and cell-penetrant small molecules (molecular weight ∼ 380 Da) that compete with the peptide docking motif for binding to PDK1. We solved the first high-resolution structure of a peptide docking motif (PIFtide) bound to PDK1 and mapped binding energy hot spots using mutational analysis. We then solved structures of PDK1 bound to the allosteric small molecules, which revealed a binding mode that remarkably mimics three of five hot-spot residues in PIFtide. These allosteric small molecules are substrate-selective PDK1 inhibitors when used as single agents, but when combined with an ATP-competitive inhibitor, they completely suppress the activation of the downstream kinases. This work provides a promising new scaffold for the development of high-affinity PIF pocket ligands, which may be used to enhance the anticancer activity of existing PDK1 inhibitors. Moreover, our results provide further impetus for exploring the helix αC patches of other protein kinases as potential therapeutic targets even though they involve protein-protein interfaces.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
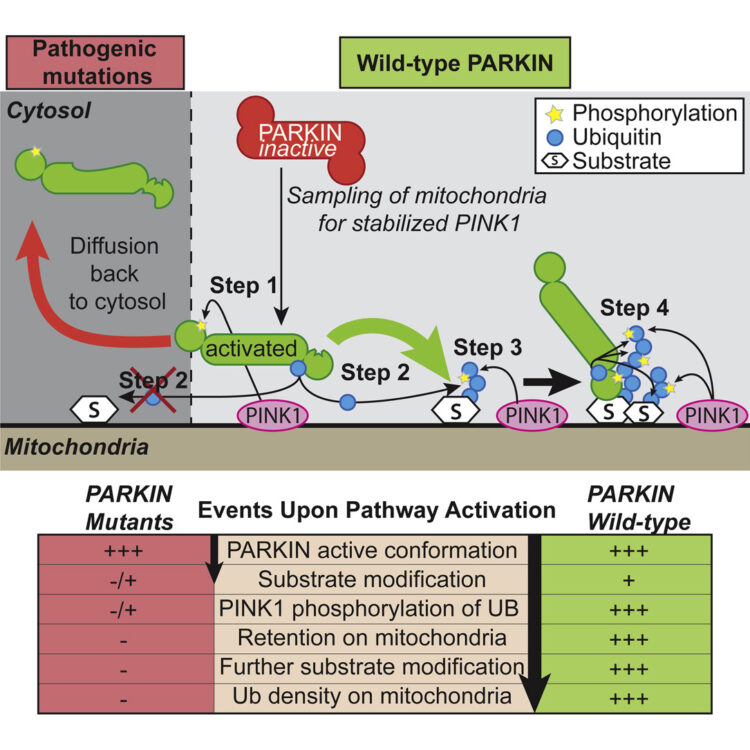
Ordureau, Alban; Sarraf, Shireen A; Duda, David M; Heo, Jin-Mi; Jedrychowski, Mark P; Sviderskiy, Vladislav O; Olszewski, Jennifer L; Koerber, James T; Xie, Tiao; Beausoleil, Sean A; Wells, James A; Gygi, Steven P; Schulman, Brenda A; Harper, J Wade
Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis Journal Article
In: Mol Cell, vol. 56, no. 3, pp. 360–375, 2014, ISSN: 1097-4164.
@article{pmid25284222,
title = {Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis},
author = {Alban Ordureau and Shireen A Sarraf and David M Duda and Jin-Mi Heo and Mark P Jedrychowski and Vladislav O Sviderskiy and Jennifer L Olszewski and James T Koerber and Tiao Xie and Sean A Beausoleil and James A Wells and Steven P Gygi and Brenda A Schulman and J Wade Harper},
doi = {10.1016/j.molcel.2014.09.007},
issn = {1097-4164},
year = {2014},
date = {2014-11-01},
urldate = {2014-11-01},
journal = {Mol Cell},
volume = {56},
number = {3},
pages = {360--375},
abstract = {Phosphorylation is often used to promote protein ubiquitylation, yet we rarely understand quantitatively how ligase activation and ubiquitin (UB) chain assembly are integrated with phosphoregulation. Here we employ quantitative proteomics and live-cell imaging to dissect individual steps in the PINK1 kinase-PARKIN UB ligase mitochondrial control pathway disrupted in Parkinson's disease. PINK1 plays a dual role by phosphorylating PARKIN on its UB-like domain and poly-UB chains on mitochondria. PARKIN activation by PINK1 produces canonical and noncanonical UB chains on mitochondria, and PARKIN-dependent chain assembly is required for accumulation of poly-phospho-UB (poly-p-UB) on mitochondria. In vitro, PINK1 directly activates PARKIN's ability to assemble canonical and noncanonical UB chains and promotes association of PARKIN with both p-UB and poly-p-UB. Our data reveal a feedforward mechanism that explains how PINK1 phosphorylation of both PARKIN and poly-UB chains synthesized by PARKIN drives a program of PARKIN recruitment and mitochondrial ubiquitylation in response to mitochondrial damage.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Julien, Olivier; Kampmann, Martin; Bassik, Michael C; Zorn, Julie A; Venditto, Vincent J; Shimbo, Kazutaka; Agard, Nicholas J; Shimada, Kenichi; Rheingold, Arnold L; Stockwell, Brent R; Weissman, Jonathan S; Wells, James A
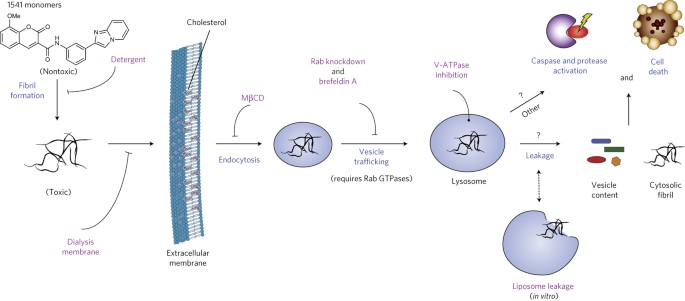
Unraveling the mechanism of cell death induced by chemical fibrils Journal Article
In: Nat Chem Biol, vol. 10, no. 11, pp. 969–976, 2014, ISSN: 1552-4469.
@article{pmid25262416,
title = {Unraveling the mechanism of cell death induced by chemical fibrils},
author = {Olivier Julien and Martin Kampmann and Michael C Bassik and Julie A Zorn and Vincent J Venditto and Kazutaka Shimbo and Nicholas J Agard and Kenichi Shimada and Arnold L Rheingold and Brent R Stockwell and Jonathan S Weissman and James A Wells},
doi = {10.1038/nchembio.1639},
issn = {1552-4469},
year = {2014},
date = {2014-11-01},
urldate = {2014-11-01},
journal = {Nat Chem Biol},
volume = {10},
number = {11},
pages = {969--976},
abstract = {We previously discovered a small-molecule inducer of cell death, named 1541, that noncovalently self-assembles into chemical fibrils ('chemi-fibrils') and activates procaspase-3 in vitro. We report here that 1541-induced cell death is caused by the fibrillar rather than the soluble form of the drug. A short hairpin RNA screen reveals that knockdown of genes involved in endocytosis, vesicle trafficking and lysosomal acidification causes partial 1541 resistance. We confirm the role of these pathways using pharmacological inhibitors. Microscopy shows that the fluorescent chemi-fibrils accumulate in punctae inside cells that partially colocalize with lysosomes. Notably, the chemi-fibrils bind and induce liposome leakage in vitro, suggesting they may do the same in cells. The chemi-fibrils induce extensive proteolysis including caspase substrates, yet modulatory profiling reveals that chemi-fibrils form a distinct class from existing inducers of cell death. The chemi-fibrils share similarities with proteinaceous fibrils and may provide insight into their mechanism of cellular toxicity.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
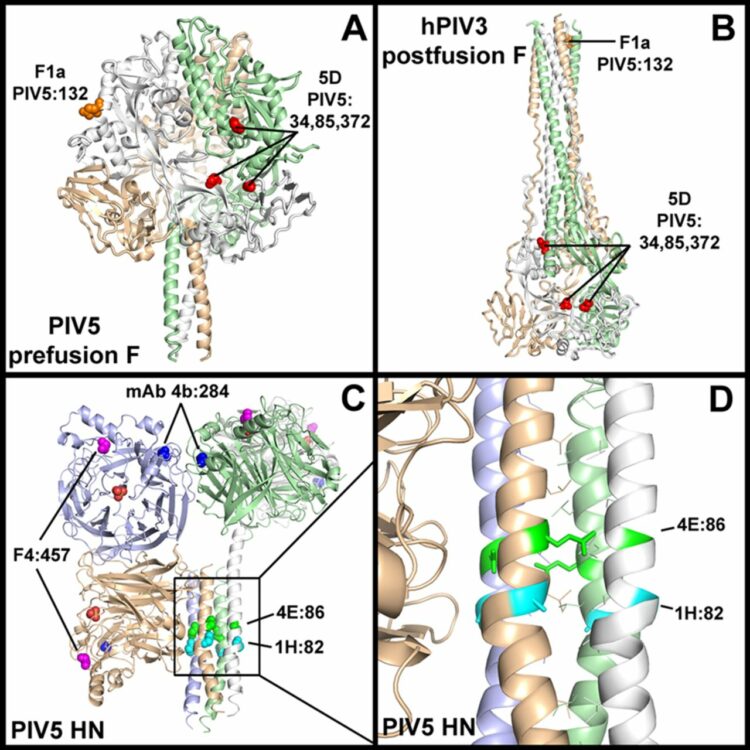
Welch, Brett D; Paduch, Marcin; Leser, George P; Bergman, Zachary; Kors, Christopher A; Paterson, Reay G; Jardetzky, Theodore S; Kossiakoff, Anthony A; Lamb, Robert A
Probing the functions of the paramyxovirus glycoproteins F and HN with a panel of synthetic antibodies Journal Article
In: J Virol, vol. 88, no. 20, pp. 11713–11725, 2014, ISSN: 1098-5514.
@article{pmid25122782,
title = {Probing the functions of the paramyxovirus glycoproteins F and HN with a panel of synthetic antibodies},
author = {Brett D Welch and Marcin Paduch and George P Leser and Zachary Bergman and Christopher A Kors and Reay G Paterson and Theodore S Jardetzky and Anthony A Kossiakoff and Robert A Lamb},
doi = {10.1128/JVI.01707-14},
issn = {1098-5514},
year = {2014},
date = {2014-10-01},
urldate = {2014-10-01},
journal = {J Virol},
volume = {88},
number = {20},
pages = {11713--11725},
abstract = {Paramyxoviruses are enveloped negative-strand RNA viruses that are significant human and animal pathogens. Most paramyxoviruses infect host cells via the concerted action of a tetrameric attachment protein (variously called HN, H, or G) that binds either sialic acid or protein receptors on target cells and a trimeric fusion protein (F) that merges the viral envelope with the plasma membrane at neutral pH. F initially folds to a metastable prefusion conformation that becomes activated via a cleavage event during cellular trafficking. Upon receptor binding, the attachment protein, which consists of a globular head anchored to the membrane via a helical tetrameric stalk, triggers a major conformation change in F which results in fusion of virus and host cell membranes. We recently proposed a model for F activation in which the attachment protein head domains move following receptor binding to expose HN stalk residues critical for triggering F. To test the model in the context of wild-type viral glycoproteins, we used a restricted-diversity combinatorial Fab library and phage display to rapidly generate synthetic antibodies (sAbs) against multiple domains of the paramyxovirus parainfluenza 5 (PIV5) pre- and postfusion F and HN. As predicted by the model, sAbs that bind to the critical F-triggering region of the HN stalk do not disrupt receptor binding or neuraminidase (NA) activity but are potent inhibitors of fusion. An inhibitory prefusion F-specific sAb recognized a quaternary antigenic site and may inhibit fusion by preventing F refolding or by blocking the F-HN interaction. Importance: The paramyxovirus family of negative-strand RNA viruses cause significant disease in humans and animals. The viruses bind to cells via their receptor binding protein and then enter cells by fusion of their envelope with the host cell plasma membrane, a process mediated by a metastable viral fusion (F) protein. To understand the steps in viral membrane fusion, a library of synthetic antibodies to F protein and the receptor binding protein was generated in bacteriophage. These antibodies bound to different regions of the F protein and the receptor binding protein, and the location of antibody binding affected different processes in viral entry into cells.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
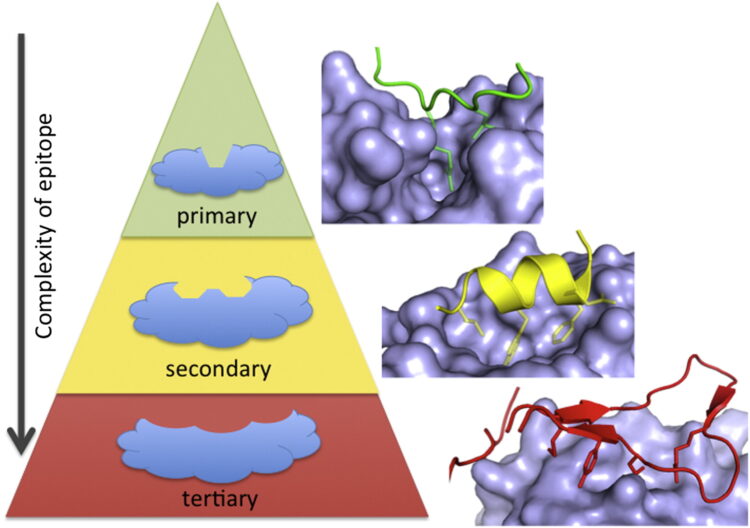
Arkin, Michelle R; Tang, Yinyan; Wells, James A
Small-molecule inhibitors of protein-protein interactions: progressing toward the reality Journal Article
In: Chem Biol, vol. 21, no. 9, pp. 1102–1114, 2014, ISSN: 1879-1301.
@article{pmid25237857,
title = {Small-molecule inhibitors of protein-protein interactions: progressing toward the reality},
author = {Michelle R Arkin and Yinyan Tang and James A Wells},
doi = {10.1016/j.chembiol.2014.09.001},
issn = {1879-1301},
year = {2014},
date = {2014-09-01},
urldate = {2014-09-01},
journal = {Chem Biol},
volume = {21},
number = {9},
pages = {1102--1114},
abstract = {The past 20 years have seen many advances in our understanding of protein-protein interactions (PPIs) and how to target them with small-molecule therapeutics. In 2004, we reviewed some early successes; since then, potent inhibitors have been developed for diverse protein complexes, and compounds are now in clinical trials for six targets. Surprisingly, many of these PPI clinical candidates have efficiency metrics typical of "lead-like" or "drug-like" molecules and are orally available. Successful discovery efforts have integrated multiple disciplines and make use of all the modern tools of target-based discovery-structure, computation, screening, and biomarkers. PPIs become progressively more challenging as the interfaces become more complex, i.e., as binding epitopes are displayed on primary, secondary, or tertiary structures. Here, we review the last 10 years of progress, focusing on the properties of PPI inhibitors that have advanced to clinical trials and prospects for the future of PPI drug discovery.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
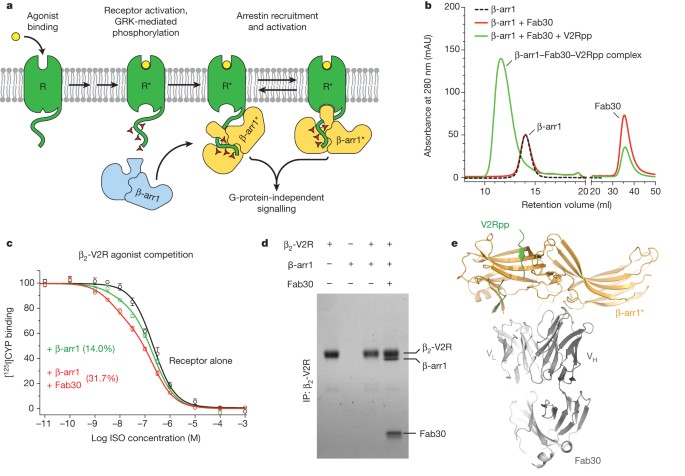
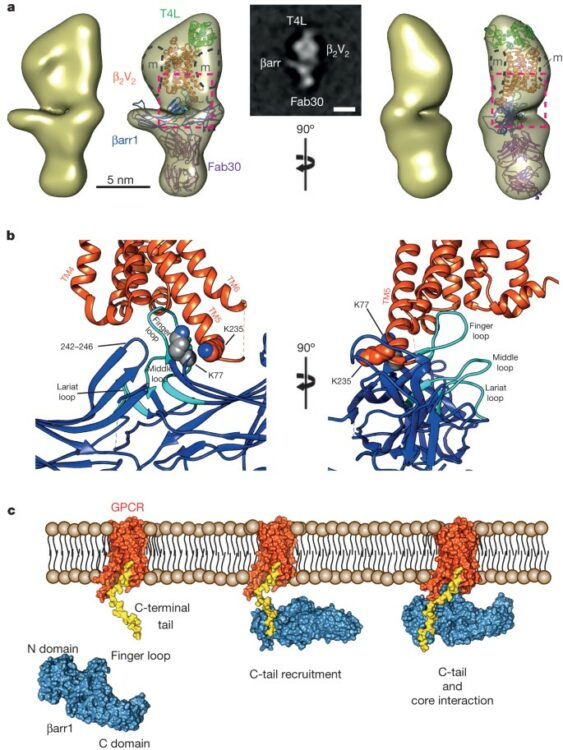
Shukla, Arun K; Westfield, Gerwin H; Xiao, Kunhong; Reis, Rosana I; Huang, Li-Yin; Tripathi-Shukla, Prachi; Qian, Jiang; Li, Sheng; Blanc, Adi; Oleskie, Austin N; Dosey, Anne M; Su, Min; Liang, Cui-Rong; Gu, Ling-Ling; Shan, Jin-Ming; Chen, Xin; Hanna, Rachel; Choi, Minjung; Yao, Xiao Jie; Klink, Bjoern U; Kahsai, Alem W; Sidhu, Sachdev S; Koide, Shohei; Penczek, Pawel A; Kossiakoff, Anthony A; Woods, Virgil L; Kobilka, Brian K; Skiniotis, Georgios; Lefkowitz, Robert J
Visualization of arrestin recruitment by a G-protein-coupled receptor Journal Article
In: Nature, vol. 512, no. 7513, pp. 218–222, 2014, ISSN: 1476-4687.
@article{pmid25043026,
title = {Visualization of arrestin recruitment by a G-protein-coupled receptor},
author = {Arun K Shukla and Gerwin H Westfield and Kunhong Xiao and Rosana I Reis and Li-Yin Huang and Prachi Tripathi-Shukla and Jiang Qian and Sheng Li and Adi Blanc and Austin N Oleskie and Anne M Dosey and Min Su and Cui-Rong Liang and Ling-Ling Gu and Jin-Ming Shan and Xin Chen and Rachel Hanna and Minjung Choi and Xiao Jie Yao and Bjoern U Klink and Alem W Kahsai and Sachdev S Sidhu and Shohei Koide and Pawel A Penczek and Anthony A Kossiakoff and Virgil L Woods and Brian K Kobilka and Georgios Skiniotis and Robert J Lefkowitz},
doi = {10.1038/nature13430},
issn = {1476-4687},
year = {2014},
date = {2014-08-01},
urldate = {2014-08-01},
journal = {Nature},
volume = {512},
number = {7513},
pages = {218--222},
abstract = {G-protein-coupled receptors (GPCRs) are critically regulated by β-arrestins, which not only desensitize G-protein signalling but also initiate a G-protein-independent wave of signalling. A recent surge of structural data on a number of GPCRs, including the β2 adrenergic receptor (β2AR)-G-protein complex, has provided novel insights into the structural basis of receptor activation. However, complementary information has been lacking on the recruitment of β-arrestins to activated GPCRs, primarily owing to challenges in obtaining stable receptor-β-arrestin complexes for structural studies. Here we devised a strategy for forming and purifying a functional human β2AR-β-arrestin-1 complex that allowed us to visualize its architecture by single-particle negative-stain electron microscopy and to characterize the interactions between β2AR and β-arrestin 1 using hydrogen-deuterium exchange mass spectrometry (HDX-MS) and chemical crosslinking. Electron microscopy two-dimensional averages and three-dimensional reconstructions reveal bimodal binding of β-arrestin 1 to the β2AR, involving two separate sets of interactions, one with the phosphorylated carboxy terminus of the receptor and the other with its seven-transmembrane core. Areas of reduced HDX together with identification of crosslinked residues suggest engagement of the finger loop of β-arrestin 1 with the seven-transmembrane core of the receptor. In contrast, focal areas of raised HDX levels indicate regions of increased dynamics in both the N and C domains of β-arrestin 1 when coupled to the β2AR. A molecular model of the β2AR-β-arrestin signalling complex was made by docking activated β-arrestin 1 and β2AR crystal structures into the electron microscopy map densities with constraints provided by HDX-MS and crosslinking, allowing us to obtain valuable insights into the overall architecture of a receptor-arrestin complex. The dynamic and structural information presented here provides a framework for better understanding the basis of GPCR regulation by arrestins.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Marcon, Edyta; Ni, Zuyao; Pu, Shuye; Turinsky, Andrei L; Trimble, Sandra Smiley; Olsen, Jonathan B; Silverman-Gavrila, Rosalind; Silverman-Gavrila, Lorelei; Phanse, Sadhna; Guo, Hongbo; Zhong, Guoqing; Guo, Xinghua; Young, Peter; Bailey, Swneke; Roudeva, Denitza; Zhao, Dorothy; Hewel, Johannes; Li, Joyce; Gräslund, Susanne; Paduch, Marcin; Kossiakoff, Anthony A; Lupien, Mathieu; Emili, Andrew; Wodak, Shoshana J; Greenblatt, Jack
Human-chromatin-related protein interactions identify a demethylase complex required for chromosome segregation Journal Article
In: Cell Rep, vol. 8, no. 1, pp. 297–310, 2014, ISSN: 2211-1247.
@article{pmid24981860,
title = {Human-chromatin-related protein interactions identify a demethylase complex required for chromosome segregation},
author = {Edyta Marcon and Zuyao Ni and Shuye Pu and Andrei L Turinsky and Sandra Smiley Trimble and Jonathan B Olsen and Rosalind Silverman-Gavrila and Lorelei Silverman-Gavrila and Sadhna Phanse and Hongbo Guo and Guoqing Zhong and Xinghua Guo and Peter Young and Swneke Bailey and Denitza Roudeva and Dorothy Zhao and Johannes Hewel and Joyce Li and Susanne Gräslund and Marcin Paduch and Anthony A Kossiakoff and Mathieu Lupien and Andrew Emili and Shoshana J Wodak and Jack Greenblatt},
doi = {10.1016/j.celrep.2014.05.050},
issn = {2211-1247},
year = {2014},
date = {2014-07-01},
urldate = {2014-07-01},
journal = {Cell Rep},
volume = {8},
number = {1},
pages = {297--310},
abstract = {Chromatin regulation is driven by multicomponent protein complexes, which form functional modules. Deciphering the components of these modules and their interactions is central to understanding the molecular pathways these proteins are regulating, their functions, and their relation to both normal development and disease. We describe the use of affinity purifications of tagged human proteins coupled with mass spectrometry to generate a protein-protein interaction map encompassing known and predicted chromatin-related proteins. On the basis of 1,394 successful purifications of 293 proteins, we report a high-confidence (85% precision) network involving 11,464 protein-protein interactions among 1,738 different human proteins, grouped into 164 often overlapping protein complexes with a particular focus on the family of JmjC-containing lysine demethylases, their partners, and their roles in chromatin remodeling. We show that RCCD1 is a partner of histone H3K36 demethylase KDM8 and demonstrate that both are important for cell-cycle-regulated transcriptional repression in centromeric regions and accurate mitotic division.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
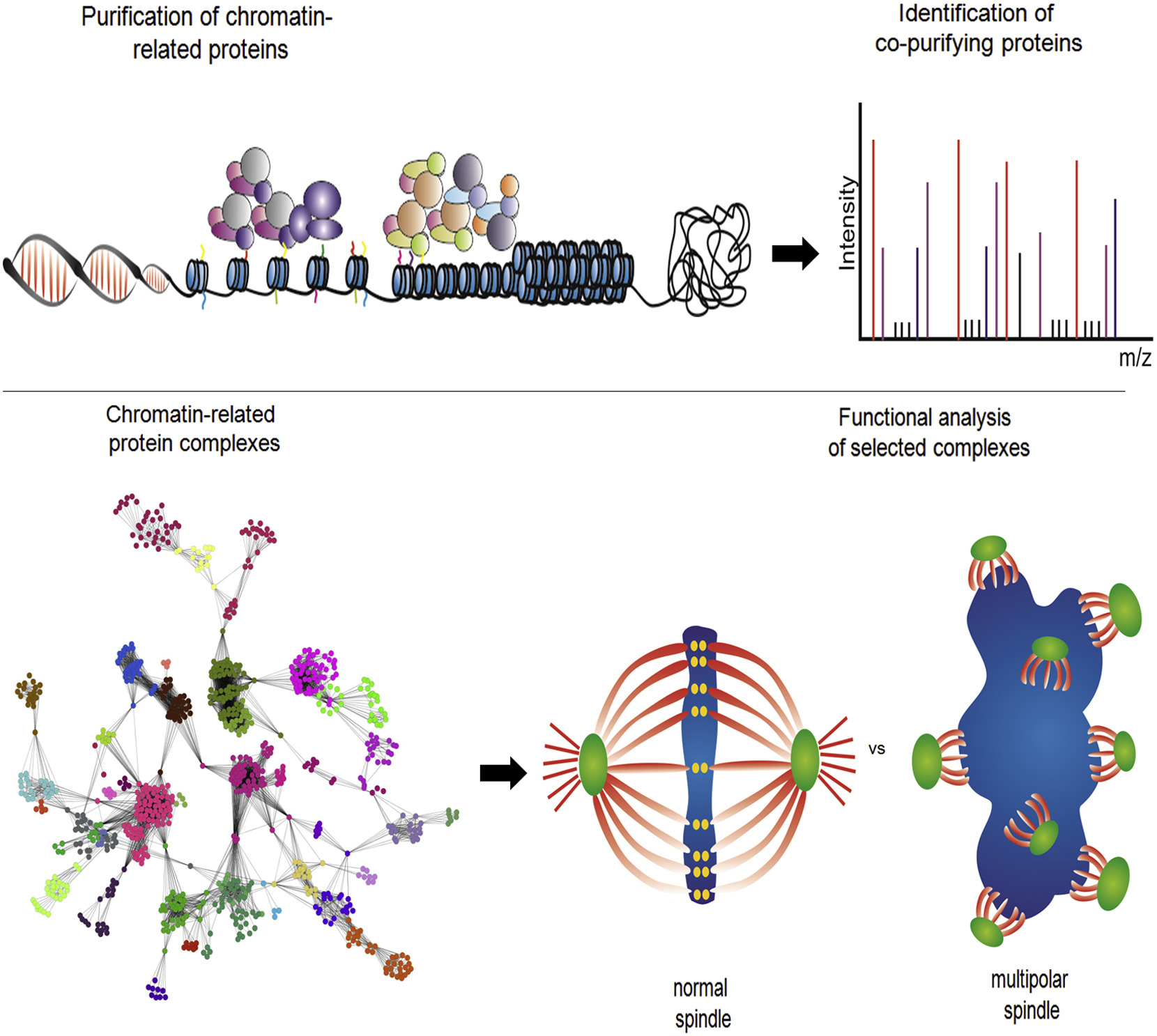
Brooks, Andrew J; Dai, Wei; O'Mara, Megan L; Abankwa, Daniel; Chhabra, Yash; Pelekanos, Rebecca A; Gardon, Olivier; Tunny, Kathryn A; Blucher, Kristopher M; Morton, Craig J; Parker, Michael W; Sierecki, Emma; Gambin, Yann; Gomez, Guillermo A; Alexandrov, Kirill; Wilson, Ian A; Doxastakis, Manolis; Mark, Alan E; Waters, Michael J
Mechanism of activation of protein kinase JAK2 by the growth hormone receptor Journal Article
In: Science, vol. 344, no. 6185, pp. 1249783, 2014, ISSN: 1095-9203.
@article{pmid24833397,
title = {Mechanism of activation of protein kinase JAK2 by the growth hormone receptor},
author = {Andrew J Brooks and Wei Dai and Megan L O'Mara and Daniel Abankwa and Yash Chhabra and Rebecca A Pelekanos and Olivier Gardon and Kathryn A Tunny and Kristopher M Blucher and Craig J Morton and Michael W Parker and Emma Sierecki and Yann Gambin and Guillermo A Gomez and Kirill Alexandrov and Ian A Wilson and Manolis Doxastakis and Alan E Mark and Michael J Waters},
doi = {10.1126/science.1249783},
issn = {1095-9203},
year = {2014},
date = {2014-05-01},
urldate = {2014-05-01},
journal = {Science},
volume = {344},
number = {6185},
pages = {1249783},
abstract = {Signaling from JAK (Janus kinase) protein kinases to STAT (signal transducers and activators of transcription) transcription factors is key to many aspects of biology and medicine, yet the mechanism by which cytokine receptors initiate signaling is enigmatic. We present a complete mechanistic model for activation of receptor-bound JAK2, based on an archetypal cytokine receptor, the growth hormone receptor. For this, we used fluorescence resonance energy transfer to monitor positioning of the JAK2 binding motif in the receptor dimer, substitution of the receptor extracellular domains with Jun zippers to control the position of its transmembrane (TM) helices, atomistic modeling of TM helix movements, and docking of the crystal structures of the JAK2 kinase and its inhibitory pseudokinase domain with an opposing kinase-pseudokinase domain pair. Activation of the receptor dimer induced a separation of its JAK2 binding motifs, driven by a ligand-induced transition from a parallel TM helix pair to a left-handed crossover arrangement. This separation leads to removal of the pseudokinase domain from the kinase domain of the partner JAK2 and pairing of the two kinase domains, facilitating trans-activation. This model may well generalize to other class I cytokine receptors.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Wells, James A; Kossiakoff, Anthony A
Cell biology. New tricks for an old dimer Journal Article
In: Science, vol. 344, no. 6185, pp. 703–704, 2014, ISSN: 1095-9203.
@article{pmid24833381,
title = {Cell biology. New tricks for an old dimer},
author = {James A Wells and Anthony A Kossiakoff},
doi = {10.1126/science.1254799},
issn = {1095-9203},
year = {2014},
date = {2014-05-01},
urldate = {2014-05-01},
journal = {Science},
volume = {344},
number = {6185},
pages = {703--704},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
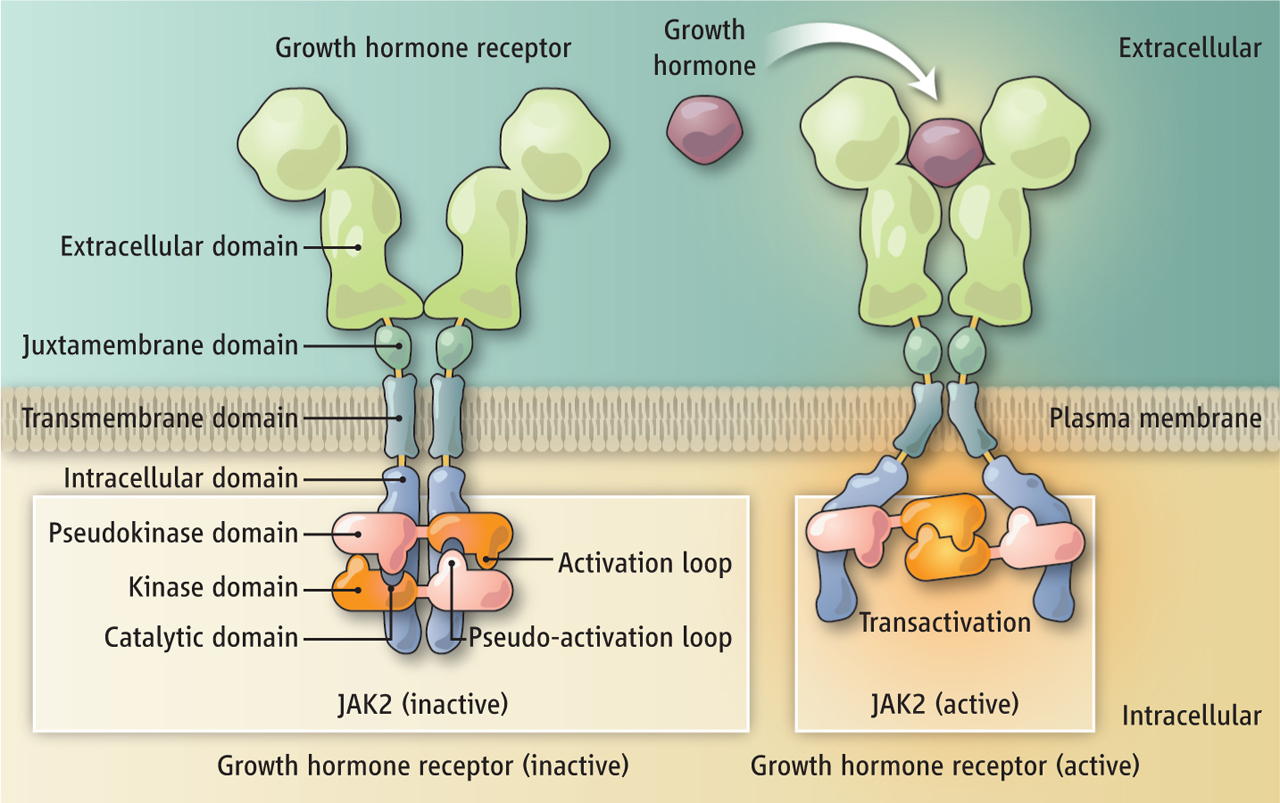
Wiita, Arun P; Hsu, Gerald W; Lu, Chuanyi M; Esensten, Jonathan H; Wells, James A
Circulating proteolytic signatures of chemotherapy-induced cell death in humans discovered by N-terminal labeling Journal Article
In: Proc Natl Acad Sci U S A, vol. 111, no. 21, pp. 7594–7599, 2014, ISSN: 1091-6490.
@article{pmid24821784,
title = {Circulating proteolytic signatures of chemotherapy-induced cell death in humans discovered by N-terminal labeling},
author = {Arun P Wiita and Gerald W Hsu and Chuanyi M Lu and Jonathan H Esensten and James A Wells},
doi = {10.1073/pnas.1405987111},
issn = {1091-6490},
year = {2014},
date = {2014-05-01},
urldate = {2014-05-01},
journal = {Proc Natl Acad Sci U S A},
volume = {111},
number = {21},
pages = {7594--7599},
abstract = {It is known that many chemotherapeutics induce cellular apoptosis over hours to days. During apoptosis, numerous cellular proteases are activated, most canonically the caspases. We speculated that detection of proteolytic fragments released from apoptotic cells into the peripheral blood may serve as a unique indicator of chemotherapy-induced cell death. Here we used an enzymatic labeling process to positively enrich free peptide α-amines in the plasma of hematologic malignancy patients soon after beginning treatment. This N-terminomic approach largely avoids interference by high-abundance proteins that complicate traditional plasma proteomic analyses. Significantly, by mass spectrometry methods, we found strong biological signatures of apoptosis directly in the postchemotherapy plasma, including numerous caspase-cleaved peptides as well as relevant peptides from apoptotic and cell-stress proteins second mitochondria-derived activator of caspases, HtrA serine peptidase 2, and activating transcription factor 6. We also treated hematologic cancer cell lines with clinically relevant chemotherapeutics and monitored proteolytic fragments released into the media. Remarkably, many of these peptides coincided with those found in patient samples. Overall, we identified 153 proteolytic peptides in postchemotherapy patient plasma as potential indicators of cellular apoptosis. Through targeted quantitative proteomics, we verified that many of these peptides were indeed increased post- vs. prechemotherapy in additional patients. Our findings reveal that numerous proteolytic fragments are released from dying tumor cells. Monitoring posttreatment proteolysis may lead to a novel class of inexpensive, rapid biomarkers of cell death.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Li, Qufei; Wanderling, Sherry; Paduch, Marcin; Medovoy, David; Singharoy, Abhishek; McGreevy, Ryan; Villalba-Galea, Carlos A; Hulse, Raymond E; Roux, Benoît; Schulten, Klaus; Kossiakoff, Anthony; Perozo, Eduardo
Structural mechanism of voltage-dependent gating in an isolated voltage-sensing domain Journal Article
In: Nat Struct Mol Biol, vol. 21, no. 3, pp. 244–252, 2014, ISSN: 1545-9985.
@article{pmid24487958,
title = {Structural mechanism of voltage-dependent gating in an isolated voltage-sensing domain},
author = {Qufei Li and Sherry Wanderling and Marcin Paduch and David Medovoy and Abhishek Singharoy and Ryan McGreevy and Carlos A Villalba-Galea and Raymond E Hulse and Benoît Roux and Klaus Schulten and Anthony Kossiakoff and Eduardo Perozo},
doi = {10.1038/nsmb.2768},
issn = {1545-9985},
year = {2014},
date = {2014-03-01},
urldate = {2014-03-01},
journal = {Nat Struct Mol Biol},
volume = {21},
number = {3},
pages = {244--252},
abstract = {The transduction of transmembrane electric fields into protein motion has an essential role in the generation and propagation of cellular signals. Voltage-sensing domains (VSDs) carry out these functions through reorientations of positive charges in the S4 helix. Here, we determined crystal structures of the Ciona intestinalis VSD (Ci-VSD) in putatively active and resting conformations. S4 undergoes an ~5-Å displacement along its main axis, accompanied by an ~60° rotation. This movement is stabilized by an exchange in countercharge partners in helices S1 and S3 that generates an estimated net charge transfer of ~1 eo. Gating charges move relative to a ''hydrophobic gasket' that electrically divides intra- and extracellular compartments. EPR spectroscopy confirms the limited nature of S4 movement in a membrane environment. These results provide an explicit mechanism for voltage sensing and set the basis for electromechanical coupling in voltage-dependent enzymes and ion channels.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
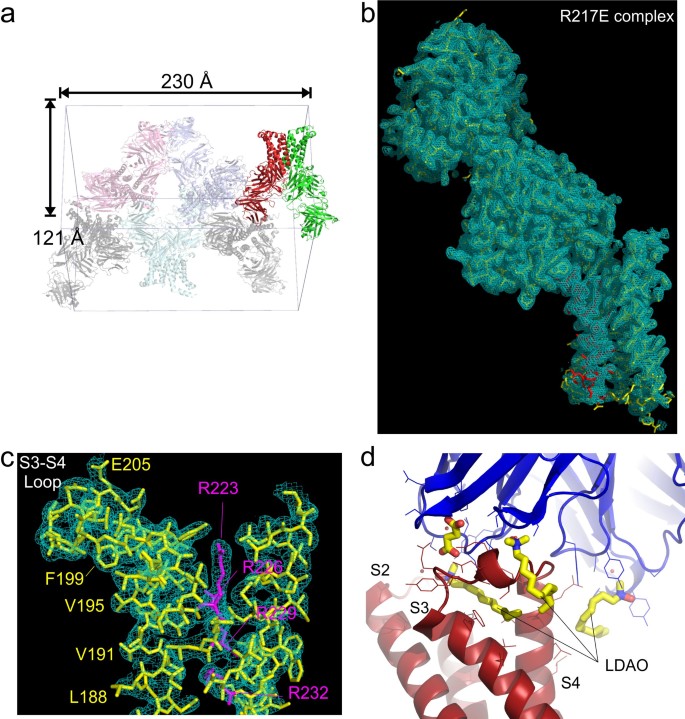
Lodge, Jean M; Rettenmaier, T Justin; Wells, James A; Pomerantz, William C; Mapp, Anna K
FP Tethering: a screening technique to rapidly identify compounds that disrupt protein-protein interactions Journal Article
In: Medchemcomm, vol. 5, pp. 370–375, 2014, ISSN: 2040-2503.
@article{pmid24795804,
title = {FP Tethering: a screening technique to rapidly identify compounds that disrupt protein-protein interactions},
author = {Jean M Lodge and T Justin Rettenmaier and James A Wells and William C Pomerantz and Anna K Mapp},
doi = {10.1039/C3MD00356F},
issn = {2040-2503},
year = {2014},
date = {2014-03-01},
urldate = {2014-03-01},
journal = {Medchemcomm},
volume = {5},
pages = {370--375},
abstract = {Tethering is a screening technique for discovering small-molecule fragments that bind to pre-determined sites via formation of a disulphide bond. Tethering screens traditionally rely upon mass spectrometry to detect disulphide bind formation, which requires a time-consuming liquid chromatography step. Here we show that Tethering can be performed rapidly and inexpensively using a homogenous fluorescence polarization (FP) assay that detects displacement of a peptide ligand from the protein target as an indirect readout of disulphide formation. We apply this method, termed FP Tethering, to identify fragments that disrupt the protein-protein interaction between the KIX domain of the transcriptional coactivator CBP and the transcriptional activator peptide pKID.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
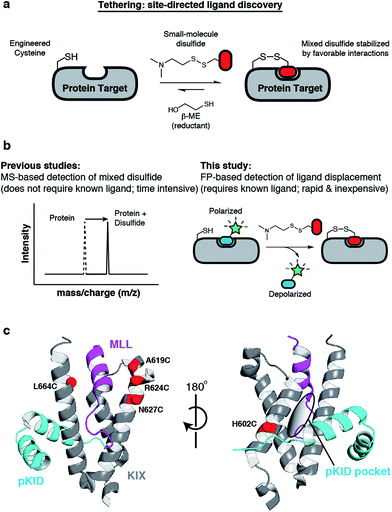
Wiita, Arun P; Seaman, Julia E; Wells, James A
Global analysis of cellular proteolysis by selective enzymatic labeling of protein N-termini Journal Article
In: Methods Enzymol, vol. 544, pp. 327–358, 2014, ISSN: 1557-7988.
@article{pmid24974296,
title = {Global analysis of cellular proteolysis by selective enzymatic labeling of protein N-termini},
author = {Arun P Wiita and Julia E Seaman and James A Wells},
doi = {10.1016/B978-0-12-417158-9.00013-3},
issn = {1557-7988},
year = {2014},
date = {2014-01-01},
urldate = {2014-01-01},
journal = {Methods Enzymol},
volume = {544},
pages = {327--358},
abstract = {Proteolysis is a critical modification leading to alteration of protein function with important outcomes in many biological processes. However, for the majority of proteases, we have an incomplete understanding of both cellular substrates and downstream effects. Here, we describe detailed protocols and applications for using the rationally engineered peptide ligase, subtiligase, to specifically label and capture protein N-termini generated by proteases either induced or added to complex biological samples. This method allows identification of the protein targets as well as their precise cleavage locations. This approach has revealed >8000 proteolytic sites in healthy and apoptotic cells including >1700 caspase cleavages. One can further determine substrate preferences through rate analysis with quantitative mass spectrometry, physiological substrate specificities, and even infer the identity of proteases operating in the cell. In this chapter, we also describe how this experimental method can be generalized to investigate proteolysis in any biological sample.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
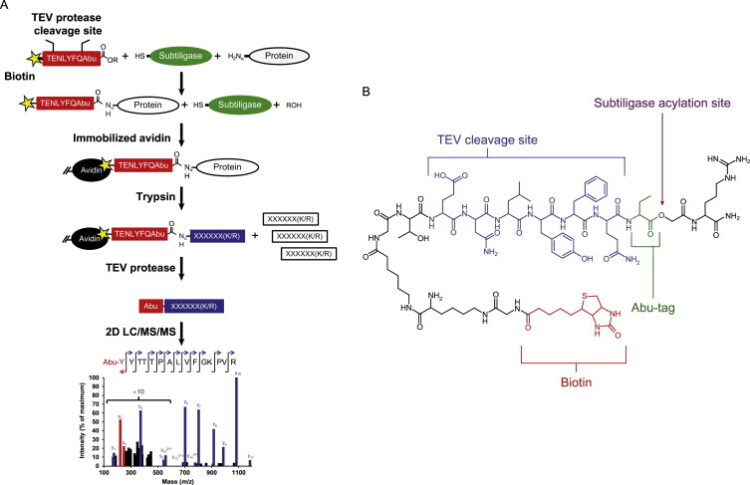
Morgan, Charles W; Julien, Olivier; Unger, Elizabeth K; Shah, Nirao M; Wells, James A
Turning on caspases with genetics and small molecules Journal Article
In: Methods Enzymol, vol. 544, pp. 179–213, 2014, ISSN: 1557-7988.
@article{pmid24974291,
title = {Turning on caspases with genetics and small molecules},
author = {Charles W Morgan and Olivier Julien and Elizabeth K Unger and Nirao M Shah and James A Wells},
doi = {10.1016/B978-0-12-417158-9.00008-X},
issn = {1557-7988},
year = {2014},
date = {2014-01-01},
urldate = {2014-01-01},
journal = {Methods Enzymol},
volume = {544},
pages = {179--213},
abstract = {Caspases, aspartate-specific cysteine proteases, have fate-determining roles in many cellular processes including apoptosis, differentiation, neuronal remodeling, and inflammation (for review, see Yuan & Kroemer, 2010). There are a dozen caspases in humans alone, yet their individual contributions toward these phenotypes are not well understood. Thus, there has been considerable interest in activating individual caspases or using their activity to drive these processes in cells and animals. We envision that such experimental control of caspase activity can not only afford novel insights into fundamental biological problems but may also enable new models for disease and suggest possible routes to therapeutic intervention. In particular, localized, genetic, and small-molecule-controlled caspase activation has the potential to target the desired cell type in a tissue. Suppression of caspase activation is one of the hallmarks of cancer and thus there has been significant enthusiasm for generating selective small-molecule activators that could bypass upstream mutational events that prevent apoptosis. Here, we provide a practical guide that investigators have devised, using genetics or small molecules, to activate specific caspases in cells or animals. Additionally, we show genetically controlled activation of an executioner caspase to target the function of a defined group of neurons in the adult mammalian brain.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
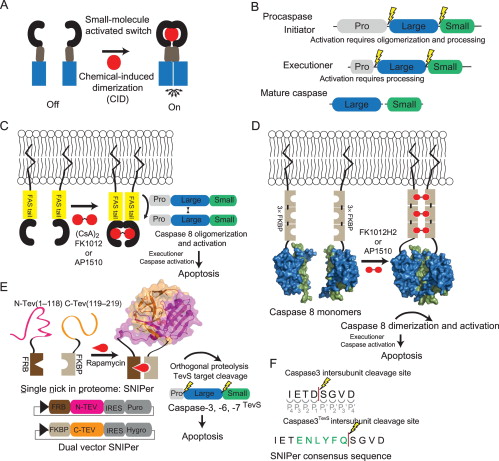
Ostrem, Jonathan M; Peters, Ulf; Sos, Martin L; Wells, James A; Shokat, Kevan M
K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions Journal Article
In: Nature, vol. 503, no. 7477, pp. 548–551, 2013, ISSN: 1476-4687.
@article{pmid24256730,
title = {K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions},
author = {Jonathan M Ostrem and Ulf Peters and Martin L Sos and James A Wells and Kevan M Shokat},
doi = {10.1038/nature12796},
issn = {1476-4687},
year = {2013},
date = {2013-11-01},
urldate = {2013-11-01},
journal = {Nature},
volume = {503},
number = {7477},
pages = {548--551},
abstract = {Somatic mutations in the small GTPase K-Ras are the most common activating lesions found in human cancer, and are generally associated with poor response to standard therapies. Efforts to target this oncogene directly have faced difficulties owing to its picomolar affinity for GTP/GDP and the absence of known allosteric regulatory sites. Oncogenic mutations result in functional activation of Ras family proteins by impairing GTP hydrolysis. With diminished regulation by GTPase activity, the nucleotide state of Ras becomes more dependent on relative nucleotide affinity and concentration. This gives GTP an advantage over GDP and increases the proportion of active GTP-bound Ras. Here we report the development of small molecules that irreversibly bind to a common oncogenic mutant, K-Ras(G12C). These compounds rely on the mutant cysteine for binding and therefore do not affect the wild-type protein. Crystallographic studies reveal the formation of a new pocket that is not apparent in previous structures of Ras, beneath the effector binding switch-II region. Binding of these inhibitors to K-Ras(G12C) disrupts both switch-I and switch-II, subverting the native nucleotide preference to favour GDP over GTP and impairing binding to Raf. Our data provide structure-based validation of a new allosteric regulatory site on Ras that is targetable in a mutant-specific manner.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Wiita, Arun P; Ziv, Etay; Wiita, Paul J; Urisman, Anatoly; Julien, Olivier; Burlingame, Alma L; Weissman, Jonathan S; Wells, James A
Global cellular response to chemotherapy-induced apoptosis Journal Article
In: Elife, vol. 2, pp. e01236, 2013, ISSN: 2050-084X.
@article{pmid24171104,
title = {Global cellular response to chemotherapy-induced apoptosis},
author = {Arun P Wiita and Etay Ziv and Paul J Wiita and Anatoly Urisman and Olivier Julien and Alma L Burlingame and Jonathan S Weissman and James A Wells},
doi = {10.7554/eLife.01236},
issn = {2050-084X},
year = {2013},
date = {2013-10-01},
urldate = {2013-10-01},
journal = {Elife},
volume = {2},
pages = {e01236},
abstract = {How cancer cells globally struggle with a chemotherapeutic insult before succumbing to apoptosis is largely unknown. Here we use an integrated systems-level examination of transcription, translation, and proteolysis to understand these events central to cancer treatment. As a model we study myeloma cells exposed to the proteasome inhibitor bortezomib, a first-line therapy. Despite robust transcriptional changes, unbiased quantitative proteomics detects production of only a few critical anti-apoptotic proteins against a background of general translation inhibition. Simultaneous ribosome profiling further reveals potential translational regulation of stress response genes. Once the apoptotic machinery is engaged, degradation by caspases is largely independent of upstream bortezomib effects. Moreover, previously uncharacterized non-caspase proteolytic events also participate in cellular deconstruction. Our systems-level data also support co-targeting the anti-apoptotic regulator HSF1 to promote cell death by bortezomib. This integrated approach offers unique, in-depth insight into apoptotic dynamics that may prove important to preclinical evaluation of any anti-cancer compound. DOI:http://dx.doi.org/10.7554/eLife.01236.001.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

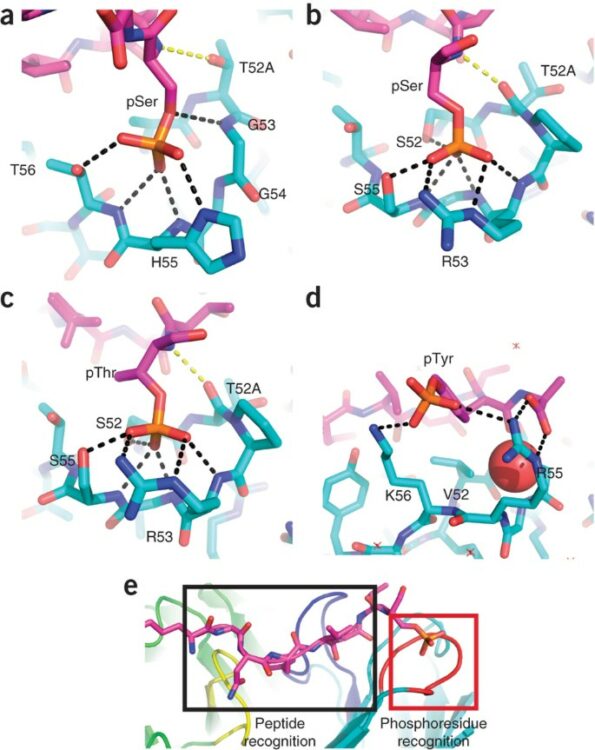
Koerber, James T; Thomsen, Nathan D; Hannigan, Brett T; Degrado, William F; Wells, James A
Nature-inspired design of motif-specific antibody scaffolds Journal Article
In: Nat Biotechnol, vol. 31, no. 10, pp. 916–921, 2013, ISSN: 1546-1696.
@article{pmid23955275,
title = {Nature-inspired design of motif-specific antibody scaffolds},
author = {James T Koerber and Nathan D Thomsen and Brett T Hannigan and William F Degrado and James A Wells},
doi = {10.1038/nbt.2672},
issn = {1546-1696},
year = {2013},
date = {2013-10-01},
urldate = {2013-10-01},
journal = {Nat Biotechnol},
volume = {31},
number = {10},
pages = {916--921},
abstract = {Aberrant changes in post-translational modifications (PTMs) such as phosphate groups underlie a majority of human diseases. However, detection and quantification of PTMs for diagnostic or biomarker applications often require PTM-specific monoclonal antibodies (mAbs), which are challenging to generate using traditional antibody-selection methods. Here we outline a general strategy for producing synthetic, PTM-specific mAbs by engineering a motif-specific 'hot spot' into an antibody scaffold. Inspired by a natural phosphate-binding motif, we designed and selected mAb scaffolds with hot spots specific for phosphoserine, phosphothreonine or phosphotyrosine. Crystal structures of the phospho-specific mAbs revealed two distinct modes of phosphoresidue recognition. Our data suggest that each hot spot functions independently of the surrounding scaffold, as phage display antibody libraries using these scaffolds yielded >50 phospho- and target-specific mAbs against 70% of target peptides. Our motif-specific scaffold strategy may provide a general solution for rapid, robust development of anti-PTM mAbs for signaling, diagnostic and therapeutic applications.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Carter, Paul J; Hazuda, Daria; Wells, James A
Next generation therapeutics Miscellaneous
2013, ISSN: 1879-0402.
@misc{pmid23683350,
title = {Next generation therapeutics},
author = {Paul J Carter and Daria Hazuda and James A Wells},
doi = {10.1016/j.cbpa.2013.04.022},
issn = {1879-0402},
year = {2013},
date = {2013-06-01},
journal = {Curr Opin Chem Biol},
volume = {17},
number = {3},
pages = {317--319},
keywords = {},
pubstate = {published},
tppubtype = {misc}
}
Shukla, Arun K; Manglik, Aashish; Kruse, Andrew C; Xiao, Kunhong; Reis, Rosana I; Tseng, Wei-Chou; Staus, Dean P; Hilger, Daniel; Uysal, Serdar; Huang, Li-Yin; Paduch, Marcin; Tripathi-Shukla, Prachi; Koide, Akiko; Koide, Shohei; Weis, William I; Kossiakoff, Anthony A; Kobilka, Brian K; Lefkowitz, Robert J
Structure of active β-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide Journal Article
In: Nature, vol. 497, no. 7447, pp. 137–141, 2013, ISSN: 1476-4687.
@article{pmid23604254,
title = {Structure of active β-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide},
author = {Arun K Shukla and Aashish Manglik and Andrew C Kruse and Kunhong Xiao and Rosana I Reis and Wei-Chou Tseng and Dean P Staus and Daniel Hilger and Serdar Uysal and Li-Yin Huang and Marcin Paduch and Prachi Tripathi-Shukla and Akiko Koide and Shohei Koide and William I Weis and Anthony A Kossiakoff and Brian K Kobilka and Robert J Lefkowitz},
doi = {10.1038/nature12120},
issn = {1476-4687},
year = {2013},
date = {2013-05-01},
urldate = {2013-05-01},
journal = {Nature},
volume = {497},
number = {7447},
pages = {137--141},
abstract = {The functions of G-protein-coupled receptors (GPCRs) are primarily mediated and modulated by three families of proteins: the heterotrimeric G proteins, the G-protein-coupled receptor kinases (GRKs) and the arrestins. G proteins mediate activation of second-messenger-generating enzymes and other effectors, GRKs phosphorylate activated receptors, and arrestins subsequently bind phosphorylated receptors and cause receptor desensitization. Arrestins activated by interaction with phosphorylated receptors can also mediate G-protein-independent signalling by serving as adaptors to link receptors to numerous signalling pathways. Despite their central role in regulation and signalling of GPCRs, a structural understanding of β-arrestin activation and interaction with GPCRs is still lacking. Here we report the crystal structure of β-arrestin-1 (also called arrestin-2) in complex with a fully phosphorylated 29-amino-acid carboxy-terminal peptide derived from the human V2 vasopressin receptor (V2Rpp). This peptide has previously been shown to functionally and conformationally activate β-arrestin-1 (ref. 5). To capture this active conformation, we used a conformationally selective synthetic antibody fragment (Fab30) that recognizes the phosphopeptide-activated state of β-arrestin-1. The structure of the β-arrestin-1-V2Rpp-Fab30 complex shows marked conformational differences in β-arrestin-1 compared to its inactive conformation. These include rotation of the amino- and carboxy-terminal domains relative to each other, and a major reorientation of the 'lariat loop' implicated in maintaining the inactive state of β-arrestin-1. These results reveal, at high resolution, a receptor-interacting interface on β-arrestin, and they indicate a potentially general molecular mechanism for activation of these multifunctional signalling and regulatory proteins.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}