Publications
View by year
- Publications: 2025
- Publications: 2024
- Publications: 2023
- Publications: 2022
- Publications: 2021
- Publications: 2020
- Publications: 2019
- Publications: 2018
- Publications: 2017
- Publications: 2016
- Publications: 2015
- Publications: 2014
- Publications: 2013
- Publications: 2012
Search
Search for author, keywords, or any other term.
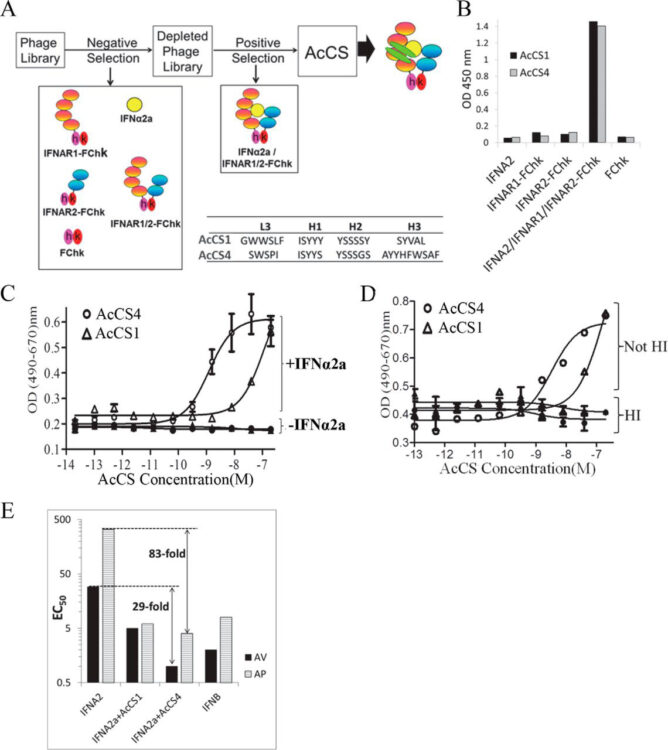
Kuruganti, Srilalitha; Miersch, Shane; Deshpande, Ashlesha; Speir, Jeffrey A; Harris, Bethany D; Schriewer, Jill M; Buller, R Mark L; Sidhu, Sachdev S; Walter, Mark R
Cytokine Activation by Antibody Fragments Targeted to Cytokine-Receptor Signaling Complexes Journal Article
In: J Biol Chem, vol. 291, no. 1, pp. 447–461, 2016, ISSN: 1083-351X.
@article{pmid26546677,
title = {Cytokine Activation by Antibody Fragments Targeted to Cytokine-Receptor Signaling Complexes},
author = {Srilalitha Kuruganti and Shane Miersch and Ashlesha Deshpande and Jeffrey A Speir and Bethany D Harris and Jill M Schriewer and R Mark L Buller and Sachdev S Sidhu and Mark R Walter},
doi = {10.1074/jbc.M115.665943},
issn = {1083-351X},
year = {2016},
date = {2016-01-01},
urldate = {2016-01-01},
journal = {J Biol Chem},
volume = {291},
number = {1},
pages = {447--461},
abstract = {Exogenous cytokine therapy can induce systemic toxicity, which might be prevented by activating endogenously produced cytokines in local cell niches. Here we developed antibody-based activators of cytokine signaling (AcCS), which recognize cytokines only when they are bound to their cell surface receptors. AcCS were developed for type I interferons (IFNs), which induce cellular activities by binding to cell surface receptors IFNAR1 and IFNAR2. As a potential alternative to exogenous IFN therapy, AcCS were shown to potentiate the biological activities of natural IFNs by ∼100-fold. Biochemical and structural characterization demonstrates that the AcCS stabilize the IFN-IFNAR2 binary complex by recognizing an IFN-induced conformational change in IFNAR2. Using IFN mutants that disrupt IFNAR1 binding, AcCS were able to enhance IFN antiviral potency without activating antiproliferative responses. This suggests AcCS can be used to manipulate cytokine signaling for basic science and possibly for therapeutic applications.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Huang, Haiming; Economopoulos, Nicolas O; Liu, Bernard A; Uetrecht, Andrea; Gu, Jun; Jarvik, Nick; Nadeem, Vincent; Pawson, Tony; Moffat, Jason; Miersch, Shane; Sidhu, Sachdev S
Selection of recombinant anti-SH3 domain antibodies by high-throughput phage display Journal Article
In: Protein Sci, vol. 24, no. 11, pp. 1890–1900, 2015, ISSN: 1469-896X.
@article{pmid26332758,
title = {Selection of recombinant anti-SH3 domain antibodies by high-throughput phage display},
author = {Haiming Huang and Nicolas O Economopoulos and Bernard A Liu and Andrea Uetrecht and Jun Gu and Nick Jarvik and Vincent Nadeem and Tony Pawson and Jason Moffat and Shane Miersch and Sachdev S Sidhu},
doi = {10.1002/pro.2799},
issn = {1469-896X},
year = {2015},
date = {2015-11-01},
urldate = {2015-11-01},
journal = {Protein Sci},
volume = {24},
number = {11},
pages = {1890--1900},
abstract = {Antibodies are indispensable tools in biochemical research and play an expanding role as therapeutics. While hybridoma technology is the dominant method for antibody production, phage display is an emerging technology. Here, we developed and employed a high-throughput pipeline that enables selection of antibodies against hundreds of antigens in parallel. Binding selections using a phage-displayed synthetic antigen-binding fragment (Fab) library against 110 human SH3 domains yielded hundreds of Fabs targeting 58 antigens. Affinity assays demonstrated that representative Fabs bind tightly and specifically to their targets. Furthermore, we developed an efficient affinity maturation strategy adaptable to high-throughput, which increased affinity dramatically but did not compromise specificity. Finally, we tested Fabs in common cell biology applications and confirmed recognition of the full-length antigen in immunoprecipitation, immunoblotting and immunofluorescence assays. In summary, we have established a rapid and robust high-throughput methodology that can be applied to generate highly functional and renewable antibodies targeting protein domains on a proteome-wide scale.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
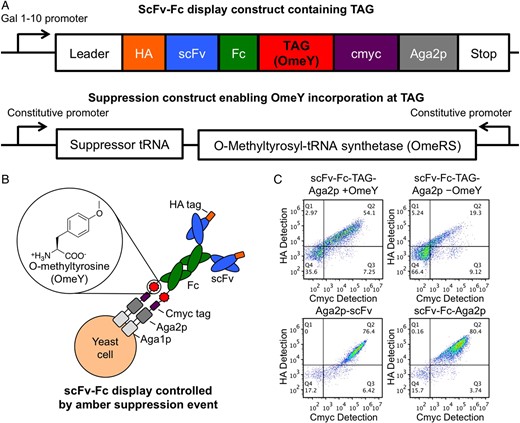
Deventer, James A Van; Kelly, Ryan L; Rajan, Saravanan; Wittrup, K Dane; Sidhu, Sachdev S
A switchable yeast display/secretion system Journal Article
In: Protein Eng Des Sel, vol. 28, no. 10, pp. 317–325, 2015, ISSN: 1741-0134.
@article{pmid26333274,
title = {A switchable yeast display/secretion system},
author = {James A Van Deventer and Ryan L Kelly and Saravanan Rajan and K Dane Wittrup and Sachdev S Sidhu},
doi = {10.1093/protein/gzv043},
issn = {1741-0134},
year = {2015},
date = {2015-10-01},
urldate = {2015-10-01},
journal = {Protein Eng Des Sel},
volume = {28},
number = {10},
pages = {317--325},
abstract = {Display technologies such as yeast and phage display offer powerful alternatives to traditional immunization-based antibody discovery, but require conversion of displayed proteins into soluble form prior to downstream characterization. Here we utilize amber suppression to implement a yeast-based switchable display/secretion system that enables the immediate production of soluble, antibody-like reagents at the end of screening efforts. Model selections in the switchable format remain efficient, and library screening in the switchable format yields renewable sources of affinity reagents exhibiting nanomolar binding affinities. These results confirm that this system provides a seamless link between display-based screening and the production and evaluation of soluble forms of candidate binding proteins. Switchable display/secretion libraries provide a cloning-free, accessible approach to affinity reagent generation.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
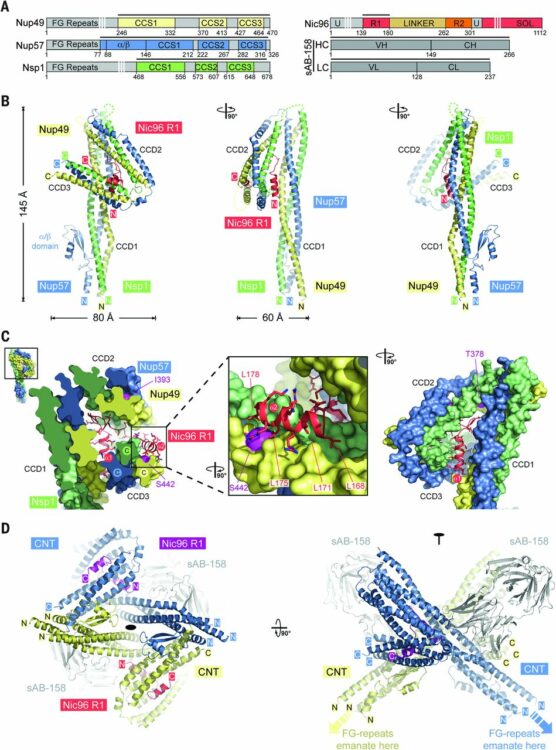
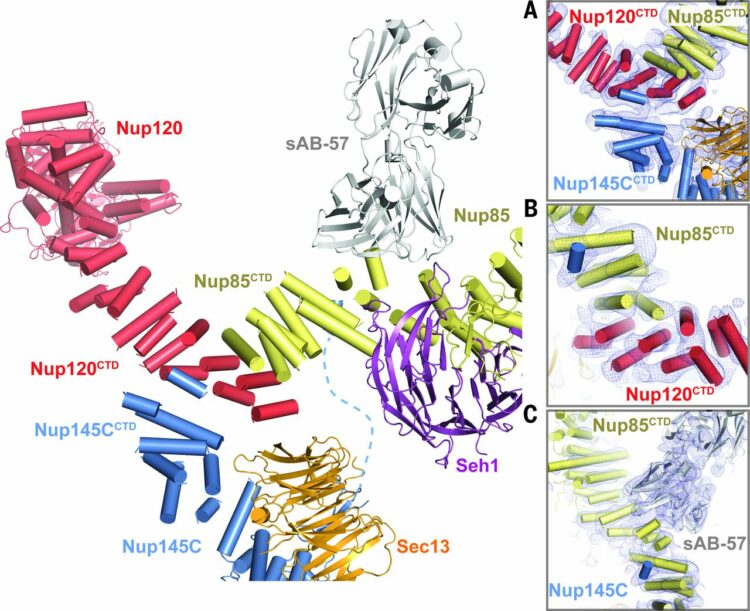
Stuwe, Tobias; Bley, Christopher J; Thierbach, Karsten; Petrovic, Stefan; Schilbach, Sandra; Mayo, Daniel J; Perriches, Thibaud; Rundlet, Emily J; Jeon, Young E; Collins, Leslie N; Huber, Ferdinand M; Lin, Daniel H; Paduch, Marcin; Koide, Akiko; Lu, Vincent; Fischer, Jessica; Hurt, Ed; Koide, Shohei; Kossiakoff, Anthony A; Hoelz, André
Architecture of the fungal nuclear pore inner ring complex Journal Article
In: Science, vol. 350, no. 6256, pp. 56–64, 2015, ISSN: 1095-9203.
@article{pmid26316600,
title = {Architecture of the fungal nuclear pore inner ring complex},
author = {Tobias Stuwe and Christopher J Bley and Karsten Thierbach and Stefan Petrovic and Sandra Schilbach and Daniel J Mayo and Thibaud Perriches and Emily J Rundlet and Young E Jeon and Leslie N Collins and Ferdinand M Huber and Daniel H Lin and Marcin Paduch and Akiko Koide and Vincent Lu and Jessica Fischer and Ed Hurt and Shohei Koide and Anthony A Kossiakoff and André Hoelz},
doi = {10.1126/science.aac9176},
issn = {1095-9203},
year = {2015},
date = {2015-10-01},
urldate = {2015-10-01},
journal = {Science},
volume = {350},
number = {6256},
pages = {56--64},
abstract = {The nuclear pore complex (NPC) constitutes the sole gateway for bidirectional nucleocytoplasmic transport. We present the reconstitution and interdisciplinary analyses of the ~425-kilodalton inner ring complex (IRC), which forms the central transport channel and diffusion barrier of the NPC, revealing its interaction network and equimolar stoichiometry. The Nsp1•Nup49•Nup57 channel nucleoporin heterotrimer (CNT) attaches to the IRC solely through the adaptor nucleoporin Nic96. The CNT•Nic96 structure reveals that Nic96 functions as an assembly sensor that recognizes the three-dimensional architecture of the CNT, thereby mediating the incorporation of a defined CNT state into the NPC. We propose that the IRC adopts a relatively rigid scaffold that recruits the CNT to primarily form the diffusion barrier of the NPC, rather than enabling channel dilation.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Hornsby, Michael; Paduch, Marcin; Miersch, Shane; Sääf, Annika; Matsuguchi, Tet; Lee, Brian; Wypisniak, Karolina; Doak, Allison; King, Daniel; Usatyuk, Svitlana; Perry, Kimberly; Lu, Vince; Thomas, William; Luke, Judy; Goodman, Jay; Hoey, Robert J; Lai, Darson; Griffin, Carly; Li, Zhijian; Vizeacoumar, Franco J; Dong, Debbie; Campbell, Elliot; Anderson, Stephen; Zhong, Nan; Gräslund, Susanne; Koide, Shohei; Moffat, Jason; Sidhu, Sachdev; Kossiakoff, Anthony; Wells, James
A High Through-put Platform for Recombinant Antibodies to Folded Proteins Journal Article
In: Mol Cell Proteomics, vol. 14, no. 10, pp. 2833–2847, 2015, ISSN: 1535-9484.
@article{pmid26290498,
title = {A High Through-put Platform for Recombinant Antibodies to Folded Proteins},
author = {Michael Hornsby and Marcin Paduch and Shane Miersch and Annika Sääf and Tet Matsuguchi and Brian Lee and Karolina Wypisniak and Allison Doak and Daniel King and Svitlana Usatyuk and Kimberly Perry and Vince Lu and William Thomas and Judy Luke and Jay Goodman and Robert J Hoey and Darson Lai and Carly Griffin and Zhijian Li and Franco J Vizeacoumar and Debbie Dong and Elliot Campbell and Stephen Anderson and Nan Zhong and Susanne Gräslund and Shohei Koide and Jason Moffat and Sachdev Sidhu and Anthony Kossiakoff and James Wells},
doi = {10.1074/mcp.O115.052209},
issn = {1535-9484},
year = {2015},
date = {2015-10-01},
urldate = {2015-10-01},
journal = {Mol Cell Proteomics},
volume = {14},
number = {10},
pages = {2833--2847},
abstract = {Antibodies are key reagents in biology and medicine, but commercial sources are rarely recombinant and thus do not provide a permanent and renewable resource. Here, we describe an industrialized platform to generate antigens and validated recombinant antibodies for 346 transcription factors (TFs) and 211 epigenetic antigens. We describe an optimized automated phage display and antigen expression pipeline that in aggregate produced about 3000 sequenced Fragment antigen-binding domain that had high affinity (typically EC50<20 nm), high stability (Tm∼80 °C), good expression in E. coli (∼5 mg/L), and ability to bind antigen in complex cell lysates. We evaluated a subset of Fabs generated to homologous SCAN domains for binding specificities. These Fragment antigen-binding domains were monospecific to their target SCAN antigen except in rare cases where they cross-reacted with a few highly related antigens. Remarkably, immunofluorescence experiments in six cell lines for 270 of the TF antigens, each having multiple antibodies, show that ∼70% stain predominantly in the cytosol and ∼20% stain in the nucleus which reinforces the dominant role that translocation plays in TF biology. These cloned antibody reagents are being made available to the academic community through our web site recombinant-antibodies.org to allow a more system-wide analysis of TF and chromatin biology. We believe these platforms, infrastructure, and automated approaches will facilitate the next generation of renewable antibody reagents to the human proteome in the coming decade.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
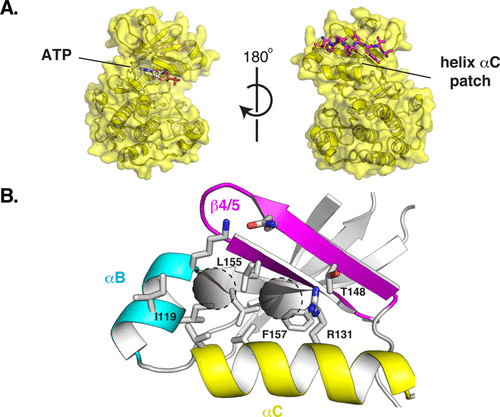
Rettenmaier, T Justin; Fan, Hao; Karpiak, Joel; Doak, Allison; Sali, Andrej; Shoichet, Brian K; Wells, James A
Small-Molecule Allosteric Modulators of the Protein Kinase PDK1 from Structure-Based Docking Journal Article
In: J Med Chem, vol. 58, no. 20, pp. 8285–8291, 2015, ISSN: 1520-4804.
@article{pmid26443011,
title = {Small-Molecule Allosteric Modulators of the Protein Kinase PDK1 from Structure-Based Docking},
author = {T Justin Rettenmaier and Hao Fan and Joel Karpiak and Allison Doak and Andrej Sali and Brian K Shoichet and James A Wells},
doi = {10.1021/acs.jmedchem.5b01216},
issn = {1520-4804},
year = {2015},
date = {2015-10-01},
urldate = {2015-10-01},
journal = {J Med Chem},
volume = {58},
number = {20},
pages = {8285--8291},
abstract = {Finding small molecules that target allosteric sites remains a grand challenge for ligand discovery. In the protein kinase field, only a handful of highly selective allosteric modulators have been found. Thus, more general methods are needed to discover allosteric modulators for additional kinases. Here, we use virtual screening against an ensemble of both crystal structures and comparative models to identify ligands for an allosteric peptide-binding site on the protein kinase PDK1 (the PIF pocket). We optimized these ligands through an analog-by-catalog search that yielded compound 4, which binds to PDK1 with 8 μM affinity. We confirmed the docking poses by determining a crystal structure of PDK1 in complex with 4. Because the PIF pocket appears to be a recurring structural feature of the kinase fold, known generally as the helix αC patch, this approach may enable the discovery of allosteric modulators for other kinases.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
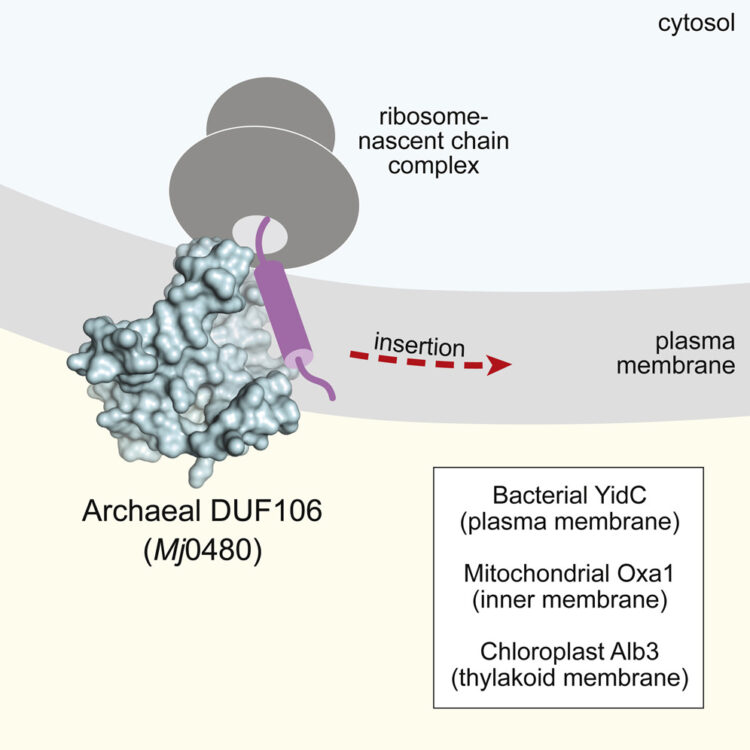
Borowska, Marta T; Dominik, Pawel K; Anghel, S Andrei; Kossiakoff, Anthony A; Keenan, Robert J
A YidC-like Protein in the Archaeal Plasma Membrane Journal Article
In: Structure, vol. 23, no. 9, pp. 1715–1724, 2015, ISSN: 1878-4186.
@article{pmid26256539,
title = {A YidC-like Protein in the Archaeal Plasma Membrane},
author = {Marta T Borowska and Pawel K Dominik and S Andrei Anghel and Anthony A Kossiakoff and Robert J Keenan},
doi = {10.1016/j.str.2015.06.025},
issn = {1878-4186},
year = {2015},
date = {2015-09-01},
urldate = {2015-09-01},
journal = {Structure},
volume = {23},
number = {9},
pages = {1715--1724},
abstract = {Cells possess specialized machinery to direct the insertion of membrane proteins into the lipid bilayer. In bacteria, the essential protein YidC inserts certain proteins into the plasma membrane, and eukaryotic orthologs are present in the mitochondrial inner membrane and the chloroplast thylakoid membrane. The existence of homologous insertases in archaea has been proposed based on phylogenetic analysis. However, limited sequence identity, distinct architecture, and the absence of experimental data have made this assignment ambiguous. Here we describe the 3.5-Å crystal structure of an archaeal DUF106 protein from Methanocaldococcus jannaschii (Mj0480), revealing a lipid-exposed hydrophilic surface presented by a conserved YidC-like fold. Functional analysis reveals selective binding of Mj0480 to ribosomes displaying a stalled YidC substrate, and a direct interaction between the buried hydrophilic surface of Mj0480 and the nascent chain. These data provide direct experimental evidence that the archaeal DUF106 proteins are YidC/Oxa1/Alb3-like insertases of the archaeal plasma membrane.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
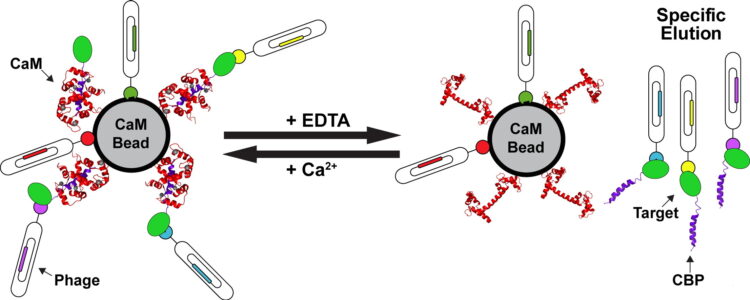
Mukherjee, Somnath; Ura, Marcin; Hoey, Robert J; Kossiakoff, Anthony A
A New Versatile Immobilization Tag Based on the Ultra High Affinity and Reversibility of the Calmodulin-Calmodulin Binding Peptide Interaction Journal Article
In: J Mol Biol, vol. 427, no. 16, pp. 2707–2725, 2015, ISSN: 1089-8638.
@article{pmid26159704,
title = {A New Versatile Immobilization Tag Based on the Ultra High Affinity and Reversibility of the Calmodulin-Calmodulin Binding Peptide Interaction},
author = {Somnath Mukherjee and Marcin Ura and Robert J Hoey and Anthony A Kossiakoff},
doi = {10.1016/j.jmb.2015.06.018},
issn = {1089-8638},
year = {2015},
date = {2015-08-01},
urldate = {2015-08-01},
journal = {J Mol Biol},
volume = {427},
number = {16},
pages = {2707--2725},
abstract = {Reversible, high-affinity immobilization tags are critical tools for myriad biological applications. However, inherent issues are associated with a number of the current methods of immobilization. Particularly, a critical element in phage display sorting is functional immobilization of target proteins. To circumvent these problems, we have used a mutant (N5A) of calmodulin binding peptide (CBP) as an immobilization tag in phage display sorting. The immobilization relies on the ultra high affinity of calmodulin to N5A mutant CBP (RWKKNFIAVSAANRFKKIS) in presence of calcium (KD~2 pM), which can be reversed by EDTA allowing controlled "capture and release" of the specific binders. To evaluate the capabilities of this system, we chose eight targets, some of which were difficult to overexpress and purify with other tags and some had failed in sorting experiments. In all cases, specific binders were generated using a Fab phage display library with CBP-fused constructs. KD values of the Fabs were in subnanomolar to low nanomolar (nM) ranges and were successfully used to selectively recognize antigens in cell-based experiments. Some of these targets were problematic even without any tag; thus, the fact that all led to successful selection endpoints means that borderline cases can be worked on with a high probability of a positive outcome. Taken together with examples of successful case specific, high-level applications like generation of conformation-, epitope- and domain-specific Fabs, we feel that the CBP tag embodies all the attributes of covalent immobilization tags but does not suffer from some of their well-documented drawbacks.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
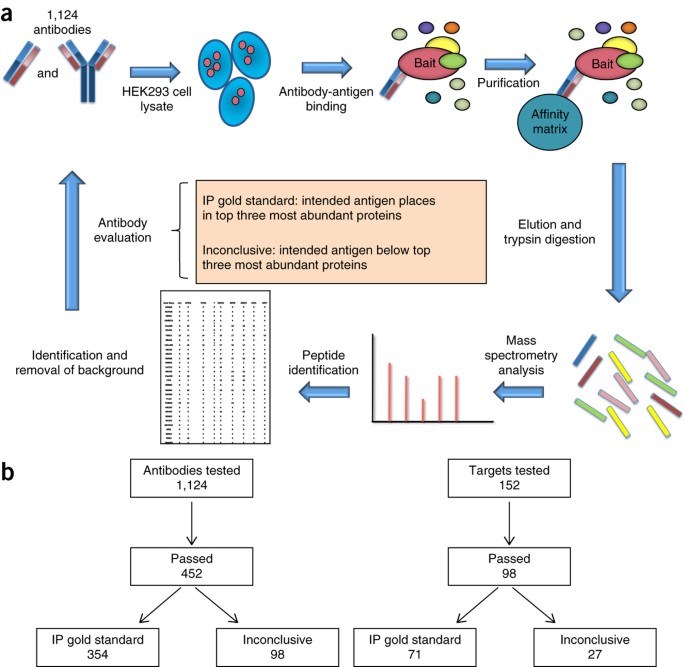
Marcon, Edyta; Jain, Harshika; Bhattacharya, Anandi; Guo, Hongbo; Phanse, Sadhna; Pu, Shuye; Byram, Gregory; Collins, Ben C; Dowdell, Evan; Fenner, Maria; Guo, Xinghua; Hutchinson, Ashley; Kennedy, Jacob J; Krastins, Bryan; Larsen, Brett; Lin, Zhen-Yuan; Lopez, Mary F; Loppnau, Peter; Miersch, Shane; Nguyen, Tin; Olsen, Jonathan B; Paduch, Marcin; Ravichandran, Mani; Seitova, Alma; Vadali, Gouri; Vogelsang, Maryann S; Whiteaker, Jeffrey R; Zhong, Guoqing; Zhong, Nan; Zhao, Lei; Aebersold, Ruedi; Arrowsmith, Cheryl H; Emili, Andrew; Frappier, Lori; Gingras, Anne-Claude; Gstaiger, Matthias; Paulovich, Amanda G; Koide, Shohei; Kossiakoff, Anthony A; Sidhu, Sachdev S; Wodak, Shoshana J; Gräslund, Susanne; Greenblatt, Jack F; Edwards, Aled M
Assessment of a method to characterize antibody selectivity and specificity for use in immunoprecipitation Journal Article
In: Nat Methods, vol. 12, no. 8, pp. 725–731, 2015, ISSN: 1548-7105.
@article{pmid26121405,
title = {Assessment of a method to characterize antibody selectivity and specificity for use in immunoprecipitation},
author = {Edyta Marcon and Harshika Jain and Anandi Bhattacharya and Hongbo Guo and Sadhna Phanse and Shuye Pu and Gregory Byram and Ben C Collins and Evan Dowdell and Maria Fenner and Xinghua Guo and Ashley Hutchinson and Jacob J Kennedy and Bryan Krastins and Brett Larsen and Zhen-Yuan Lin and Mary F Lopez and Peter Loppnau and Shane Miersch and Tin Nguyen and Jonathan B Olsen and Marcin Paduch and Mani Ravichandran and Alma Seitova and Gouri Vadali and Maryann S Vogelsang and Jeffrey R Whiteaker and Guoqing Zhong and Nan Zhong and Lei Zhao and Ruedi Aebersold and Cheryl H Arrowsmith and Andrew Emili and Lori Frappier and Anne-Claude Gingras and Matthias Gstaiger and Amanda G Paulovich and Shohei Koide and Anthony A Kossiakoff and Sachdev S Sidhu and Shoshana J Wodak and Susanne Gräslund and Jack F Greenblatt and Aled M Edwards},
doi = {10.1038/nmeth.3472},
issn = {1548-7105},
year = {2015},
date = {2015-08-01},
urldate = {2015-08-01},
journal = {Nat Methods},
volume = {12},
number = {8},
pages = {725--731},
abstract = {Antibodies are used in multiple cell biology applications, but there are no standardized methods to assess antibody quality-an absence that risks data integrity and reproducibility. We describe a mass spectrometry-based standard operating procedure for scoring immunoprecipitation antibody quality. We quantified the abundance of all the proteins in immunoprecipitates of 1,124 new recombinant antibodies for 152 chromatin-related human proteins by comparing normalized spectral abundance factors from the target antigen with those of all other proteins. We validated the performance of the standard operating procedure in blinded studies in five independent laboratories. Antibodies for which the target antigen or a member of its known protein complex was the most abundant protein were classified as 'IP gold standard'. This method generates quantitative outputs that can be stored and archived in public databases, and it represents a step toward a platform for community benchmarking of antibody quality.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
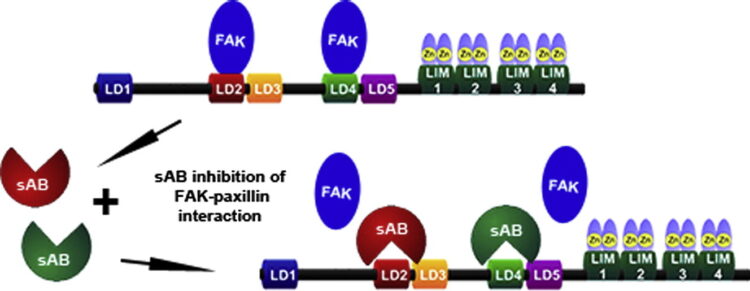
Nocula-Lugowska, Malgorzata; Lugowski, Mateusz; Salgia, Ravi; Kossiakoff, Anthony A
Engineering Synthetic Antibody Inhibitors Specific for LD2 or LD4 Motifs of Paxillin Journal Article
In: J Mol Biol, vol. 427, no. 15, pp. 2532–2547, 2015, ISSN: 1089-8638.
@article{pmid26087144,
title = {Engineering Synthetic Antibody Inhibitors Specific for LD2 or LD4 Motifs of Paxillin},
author = {Malgorzata Nocula-Lugowska and Mateusz Lugowski and Ravi Salgia and Anthony A Kossiakoff},
doi = {10.1016/j.jmb.2015.06.004},
issn = {1089-8638},
year = {2015},
date = {2015-07-01},
urldate = {2015-07-01},
journal = {J Mol Biol},
volume = {427},
number = {15},
pages = {2532--2547},
abstract = {Focal adhesion protein paxillin links integrin and growth factor signaling to actin cytoskeleton. Most of paxillin signaling activity is regulated via leucine-rich LD motifs (LD1-LD5) located at the N-terminus. Here, we demonstrate a method to engineer highly selective synthetic antibodies (sABs) against LD2 and LD4 that are binding sites for focal adhesion kinase (FAK) and other proteins. Phage display selections against peptides were used to generate sABs recognizing each LD motif. In the obtained X-ray crystal structures of the LD-sAB complexes, the LD motifs are helical and bind sABs through a hydrophobic side, similarly as in the structures with natural paxillin partners. The sABs are capable of pulling down endogenous paxillin in complex with FAK and can visualize paxillin in focal adhesions in cells. They were also used as selective inhibitors to effectively compete with focal adhesion targeting domain of FAK for the binding to LD2 and LD4. The sABs are tools for investigation of paxillin LD binding "platforms" and are capable of inhibiting paxillin interactions, thereby useful as potential therapeutics in the future.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
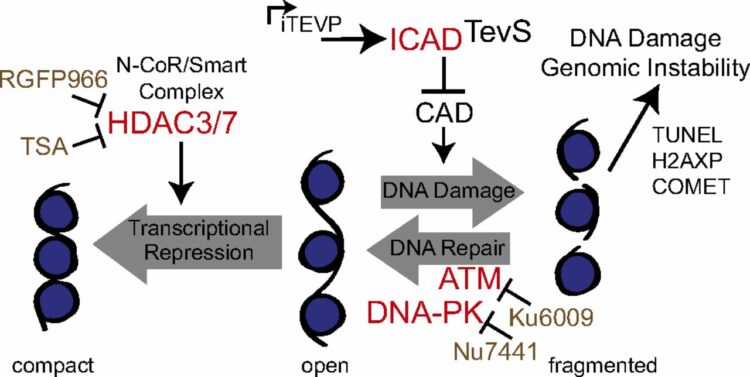
Morgan, Charles W; Diaz, Juan E; Zeitlin, Samantha G; Gray, Daniel C; Wells, James A
Engineered cellular gene-replacement platform for selective and inducible proteolytic profiling Journal Article
In: Proc Natl Acad Sci U S A, vol. 112, no. 27, pp. 8344–8349, 2015, ISSN: 1091-6490.
@article{pmid26106156,
title = {Engineered cellular gene-replacement platform for selective and inducible proteolytic profiling},
author = {Charles W Morgan and Juan E Diaz and Samantha G Zeitlin and Daniel C Gray and James A Wells},
doi = {10.1073/pnas.1504141112},
issn = {1091-6490},
year = {2015},
date = {2015-07-01},
urldate = {2015-07-01},
journal = {Proc Natl Acad Sci U S A},
volume = {112},
number = {27},
pages = {8344--8349},
abstract = {Cellular demolition during apoptosis is completed by executioner caspases, that selectively cleave more than 1,500 proteins but whose individual roles are challenging to assess. Here, we used an optimized site-specific and inducible protease to examine the role of a classic apoptotic node, the caspase-activated DNase (CAD). CAD is activated when caspases cleave its endogenous inhibitor ICAD, resulting in the characteristic DNA laddering of apoptosis. We describe a posttranscriptional gene replacement (PTGR) approach where endogenous biallelic ICAD is knocked down and simultaneously replaced with an engineered allele that is susceptible to inducible cleavage by tobacco etch virus protease. Remarkably, selective activation of CAD alone does not induce cell death, although hallmarks of DNA damage are detected in human cancer cell lines. Our data strongly support that the highly cooperative action of CAD and inhibition of DNA repair systems are critical for the DNA laddering phenotype in apoptosis. Furthermore, the PTGR approach provides a general means for replacing wild-type protein function with a precisely engineered mutant at the transcriptional level that should be useful for cell engineering studies.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Stuwe, Tobias; Correia, Ana R; Lin, Daniel H; Paduch, Marcin; Lu, Vincent T; Kossiakoff, Anthony A; Hoelz, André
Nuclear pores. Architecture of the nuclear pore complex coat Journal Article
In: Science, vol. 347, no. 6226, pp. 1148–1152, 2015, ISSN: 1095-9203.
@article{pmid25745173,
title = {Nuclear pores. Architecture of the nuclear pore complex coat},
author = {Tobias Stuwe and Ana R Correia and Daniel H Lin and Marcin Paduch and Vincent T Lu and Anthony A Kossiakoff and André Hoelz},
doi = {10.1126/science.aaa4136},
issn = {1095-9203},
year = {2015},
date = {2015-03-01},
urldate = {2015-03-01},
journal = {Science},
volume = {347},
number = {6226},
pages = {1148--1152},
abstract = {The nuclear pore complex (NPC) constitutes the sole gateway for bidirectional nucleocytoplasmic transport. Despite half a century of structural characterization, the architecture of the NPC remains unknown. Here we present the crystal structure of a reconstituted ~400-kilodalton coat nucleoporin complex (CNC) from Saccharomyces cerevisiae at a 7.4 angstrom resolution. The crystal structure revealed a curved Y-shaped architecture and the molecular details of the coat nucleoporin interactions forming the central "triskelion" of the Y. A structural comparison of the yeast CNC with an electron microscopy reconstruction of its human counterpart suggested the evolutionary conservation of the elucidated architecture. Moreover, 32 copies of the CNC crystal structure docked readily into a cryoelectron tomographic reconstruction of the fully assembled human NPC, thereby accounting for ~16 megadalton of its mass.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
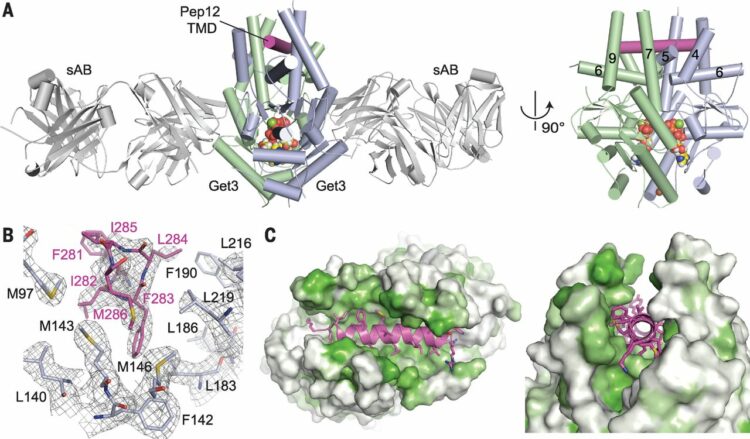
Mateja, Agnieszka; Paduch, Marcin; Chang, Hsin-Yang; Szydlowska, Anna; Kossiakoff, Anthony A; Hegde, Ramanujan S; Keenan, Robert J
Protein targeting. Structure of the Get3 targeting factor in complex with its membrane protein cargo Journal Article
In: Science, vol. 347, no. 6226, pp. 1152–1155, 2015, ISSN: 1095-9203.
@article{pmid25745174,
title = {Protein targeting. Structure of the Get3 targeting factor in complex with its membrane protein cargo},
author = {Agnieszka Mateja and Marcin Paduch and Hsin-Yang Chang and Anna Szydlowska and Anthony A Kossiakoff and Ramanujan S Hegde and Robert J Keenan},
doi = {10.1126/science.1261671},
issn = {1095-9203},
year = {2015},
date = {2015-03-01},
urldate = {2015-03-01},
journal = {Science},
volume = {347},
number = {6226},
pages = {1152--1155},
abstract = {Tail-anchored (TA) proteins are a physiologically important class of membrane proteins targeted to the endoplasmic reticulum by the conserved guided-entry of TA proteins (GET) pathway. During transit, their hydrophobic transmembrane domains (TMDs) are chaperoned by the cytosolic targeting factor Get3, but the molecular nature of the functional Get3-TA protein targeting complex remains unknown. We reconstituted the physiologic assembly pathway for a functional targeting complex and showed that it comprises a TA protein bound to a Get3 homodimer. Crystal structures of Get3 bound to different TA proteins showed an α-helical TMD occupying a hydrophobic groove that spans the Get3 homodimer. Our data elucidate the mechanism of TA protein recognition and shielding by Get3 and suggest general principles of hydrophobic domain chaperoning by cellular targeting factors.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
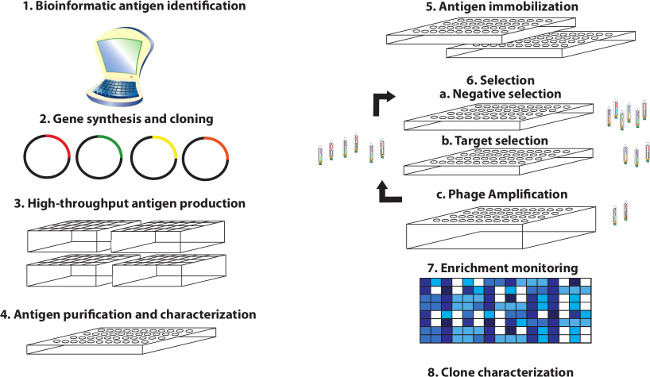
Miersch, Shane; Li, Zhijian; Hanna, Rachel; McLaughlin, Megan E; Hornsby, Michael; Matsuguchi, Tet; Paduch, Marcin; Sääf, Annika; Wells, Jim; Koide, Shohei; Kossiakoff, Anthony; Sidhu, Sachdev S
Scalable high throughput selection from phage-displayed synthetic antibody libraries Journal Article
In: J Vis Exp, no. 95, pp. 51492, 2015, ISSN: 1940-087X.
@article{pmid25651360,
title = {Scalable high throughput selection from phage-displayed synthetic antibody libraries},
author = {Shane Miersch and Zhijian Li and Rachel Hanna and Megan E McLaughlin and Michael Hornsby and Tet Matsuguchi and Marcin Paduch and Annika Sääf and Jim Wells and Shohei Koide and Anthony Kossiakoff and Sachdev S Sidhu},
doi = {10.3791/51492},
issn = {1940-087X},
year = {2015},
date = {2015-01-01},
urldate = {2015-01-01},
journal = {J Vis Exp},
number = {95},
pages = {51492},
abstract = {The demand for antibodies that fulfill the needs of both basic and clinical research applications is high and will dramatically increase in the future. However, it is apparent that traditional monoclonal technologies are not alone up to this task. This has led to the development of alternate methods to satisfy the demand for high quality and renewable affinity reagents to all accessible elements of the proteome. Toward this end, high throughput methods for conducting selections from phage-displayed synthetic antibody libraries have been devised for applications involving diverse antigens and optimized for rapid throughput and success. Herein, a protocol is described in detail that illustrates with video demonstration the parallel selection of Fab-phage clones from high diversity libraries against hundreds of targets using either a manual 96 channel liquid handler or automated robotics system. Using this protocol, a single user can generate hundreds of antigens, select antibodies to them in parallel and validate antibody binding within 6-8 weeks. Highlighted are: i) a viable antigen format, ii) pre-selection antigen characterization, iii) critical steps that influence the selection of specific and high affinity clones, and iv) ways of monitoring selection effectiveness and early stage antibody clone characterization. With this approach, we have obtained synthetic antibody fragments (Fabs) to many target classes including single-pass membrane receptors, secreted protein hormones, and multi-domain intracellular proteins. These fragments are readily converted to full-length antibodies and have been validated to exhibit high affinity and specificity. Further, they have been demonstrated to be functional in a variety of standard immunoassays including Western blotting, ELISA, cellular immunofluorescence, immunoprecipitation and related assays. This methodology will accelerate antibody discovery and ultimately bring us closer to realizing the goal of generating renewable, high quality antibodies to the proteome.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
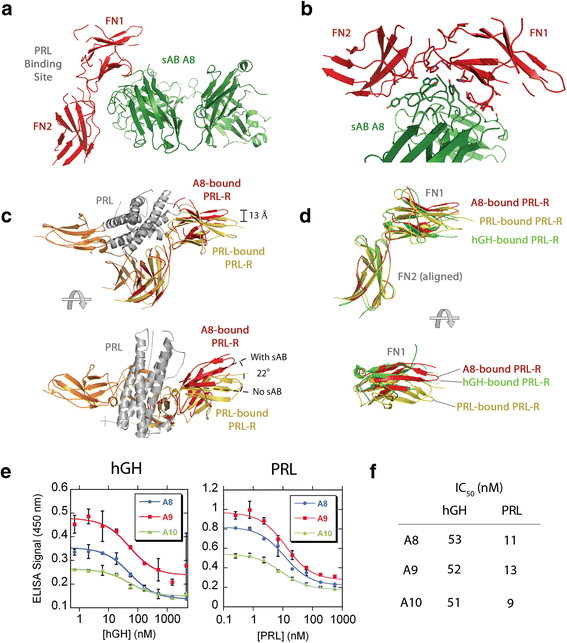
Rizk, Shahir S; Kouadio, Jean-Louis K; Szymborska, Anna; Duguid, Erica M; Mukherjee, Somnath; Zheng, Jiamao; Clevenger, Charles V; Kossiakoff, Anthony A
Engineering synthetic antibody binders for allosteric inhibition of prolactin receptor signaling Journal Article
In: Cell Commun Signal, vol. 13, pp. 1, 2015, ISSN: 1478-811X.
@article{pmid25589173,
title = {Engineering synthetic antibody binders for allosteric inhibition of prolactin receptor signaling},
author = {Shahir S Rizk and Jean-Louis K Kouadio and Anna Szymborska and Erica M Duguid and Somnath Mukherjee and Jiamao Zheng and Charles V Clevenger and Anthony A Kossiakoff},
doi = {10.1186/s12964-014-0080-8},
issn = {1478-811X},
year = {2015},
date = {2015-01-01},
urldate = {2015-01-01},
journal = {Cell Commun Signal},
volume = {13},
pages = {1},
abstract = {BACKGROUND: Many receptors function by binding to multiple ligands, each eliciting a distinct biological output. The extracellular domain of the human prolactin receptor (hPRL-R) uses an identical epitope to bind to both prolactin (hPRL) and growth hormone (hGH), yet little is known about how each hormone binding event triggers the appropriate response.nnFINDINGS: Here, we utilized a phage display library to generate synthetic antibodies (sABs) that preferentially modulate hPRL-R function in a hormone-dependent fashion. We determined the crystal structure of a sAB-hPRL-R complex, which revealed a novel allosteric mechanism of antagonism, whereby the sAB traps the receptor in a conformation more suitable for hGH binding than hPRL. This was validated by examining the effect of the sABs on hormone internalization via the hPRL-R and its downstream signaling pathway.nnCONCLUSIONS: The findings suggest that subtle structural changes in the extracellular domain of hPRL-R induced by each hormone determine the biological output triggered by hormone binding. We conclude that sABs generated by phage display selection can detect these subtle structural differences, and therefore can be used to dissect the structural basis of receptor-ligand specificity.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Zhong, Nan; Loppnau, Peter; Seitova, Alma; Ravichandran, Mani; Fenner, Maria; Jain, Harshika; Bhattacharya, Anandi; Hutchinson, Ashley; Paduch, Marcin; Lu, Vincent; Olszewski, Michal; Kossiakoff, Anthony A; Dowdell, Evan; Koide, Akiko; Koide, Shohei; Huang, Haiming; Nadeem, Vincent; Sidhu, Sachdev S; Greenblatt, Jack F; Marcon, Edyta; Arrowsmith, Cheryl H; Edwards, Aled M; Gräslund, Susanne
Optimizing Production of Antigens and Fabs in the Context of Generating Recombinant Antibodies to Human Proteins Journal Article
In: PLoS One, vol. 10, no. 10, pp. e0139695, 2015, ISSN: 1932-6203.
@article{pmid26437229,
title = {Optimizing Production of Antigens and Fabs in the Context of Generating Recombinant Antibodies to Human Proteins},
author = {Nan Zhong and Peter Loppnau and Alma Seitova and Mani Ravichandran and Maria Fenner and Harshika Jain and Anandi Bhattacharya and Ashley Hutchinson and Marcin Paduch and Vincent Lu and Michal Olszewski and Anthony A Kossiakoff and Evan Dowdell and Akiko Koide and Shohei Koide and Haiming Huang and Vincent Nadeem and Sachdev S Sidhu and Jack F Greenblatt and Edyta Marcon and Cheryl H Arrowsmith and Aled M Edwards and Susanne Gräslund},
doi = {10.1371/journal.pone.0139695},
issn = {1932-6203},
year = {2015},
date = {2015-01-01},
urldate = {2015-01-01},
journal = {PLoS One},
volume = {10},
number = {10},
pages = {e0139695},
abstract = {We developed and optimized a high-throughput project workflow to generate renewable recombinant antibodies to human proteins involved in epigenetic signalling. Three different strategies to produce phage display compatible protein antigens in bacterial systems were compared, and we found that in vivo biotinylation through the use of an Avi tag was the most productive method. Phage display selections were performed on 265 in vivo biotinylated antigen domains. High-affinity Fabs (<20nM) were obtained for 196. We constructed and optimized a new expression vector to produce in vivo biotinylated Fabs in E. coli. This increased average yields up to 10-fold, with an average yield of 4 mg/L. For 118 antigens, we identified Fabs that could immunoprecipitate their full-length endogenous targets from mammalian cell lysates. One Fab for each antigen was converted to a recombinant IgG and produced in mammalian cells, with an average yield of 15 mg/L. In summary, we have optimized each step of the pipeline to produce recombinant antibodies, significantly increasing both efficiency and yield, and also showed that these Fabs and IgGs can be generally useful for chromatin immunoprecipitation (ChIP) protocols.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
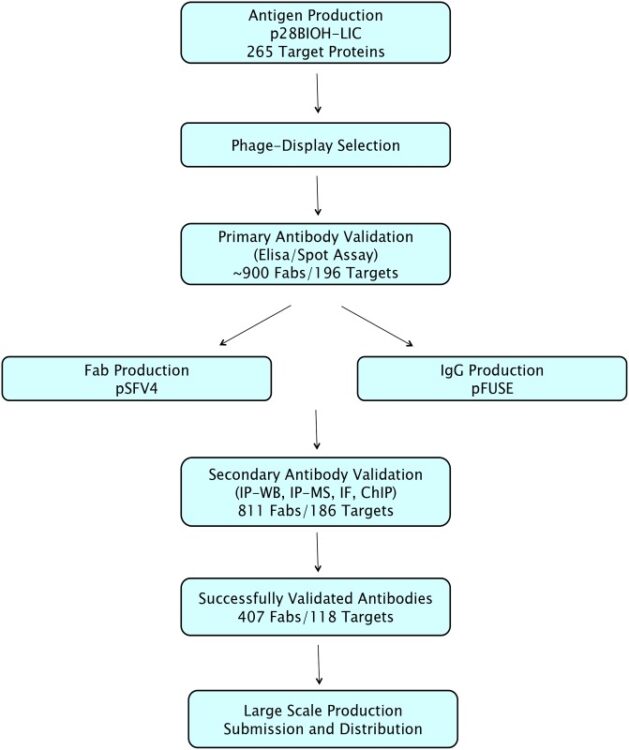
Chen, Gang; Gorelik, Leonid; Simon, Kenneth J; Pavlenco, Alevtina; Cheung, Anne; Brickelmaier, Margot; Chen, Ling Ling; Jin, Ping; Weinreb, Paul H; Sidhu, Sachdev S
In: MAbs, vol. 7, no. 4, pp. 681–692, 2015, ISSN: 1942-0870.
@article{pmid25879139,
title = {Synthetic antibodies and peptides recognizing progressive multifocal leukoencephalopathy-specific point mutations in polyomavirus JC capsid viral protein 1},
author = {Gang Chen and Leonid Gorelik and Kenneth J Simon and Alevtina Pavlenco and Anne Cheung and Margot Brickelmaier and Ling Ling Chen and Ping Jin and Paul H Weinreb and Sachdev S Sidhu},
doi = {10.1080/19420862.2015.1038447},
issn = {1942-0870},
year = {2015},
date = {2015-01-01},
urldate = {2015-01-01},
journal = {MAbs},
volume = {7},
number = {4},
pages = {681--692},
abstract = {Polyomavirus JC (JCV) is the causative agent of progressive multifocal leukoencephalopathy (PML), a rare and frequently fatal brain disease that afflicts a small fraction of the immune-compromised population, including those affected by AIDS and transplantation recipients on immunosuppressive drug therapy. Currently there is no specific therapy for PML. The major capsid viral protein 1 (VP1) involved in binding to sialic acid cell receptors is believed to be a key player in pathogenesis. PML-specific mutations in JCV VP1 sequences present at the binding pocket of sialic acid cell receptors, such as L55F and S269F, abolish sialic acid recognition and might favor PML onset. Early diagnosis of these PML-specific mutations may help identify patients at high risk of PML, thus reducing the risks associated with immunosuppressive therapy. As a first step in the development of such early diagnostic tools, we report identification and characterization of affinity reagents that specifically recognize PML-specific mutations in VP1 variants using phage display technology. We first identified 2 peptides targeting wild type VP1 with moderate specificity. Fine-tuning via selection of biased libraries designed based on 2 parental peptides yielded peptides with different, yet still moderate, bindinspecificities. In contrast, we had great success in identifying synthetic antibodies that recognize one of the PML-specific mutations (L55F) with high specificity from the phage-displayed libraries. These peptides and synthetic antibodies represent potential candidates for developing tailored immune-based assays for PML risk stratification in addition to complementing affinity reagents currently available for the study of PML and JCV.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
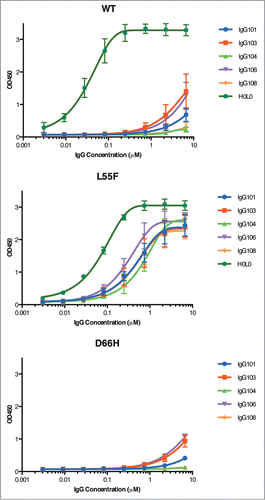
Koerber, James T; Hornsby, Michael J; Wells, James A
An improved single-chain Fab platform for efficient display and recombinant expression Journal Article
In: J Mol Biol, vol. 427, no. 2, pp. 576–586, 2015, ISSN: 1089-8638.
@article{pmid25481745,
title = {An improved single-chain Fab platform for efficient display and recombinant expression},
author = {James T Koerber and Michael J Hornsby and James A Wells},
doi = {10.1016/j.jmb.2014.11.017},
issn = {1089-8638},
year = {2015},
date = {2015-01-01},
urldate = {2015-01-01},
journal = {J Mol Biol},
volume = {427},
number = {2},
pages = {576--586},
abstract = {Antibody phage display libraries combined with high-throughput selections have recently demonstrated tremendous promise to create the next generation of renewable, recombinant antibodies to study proteins and their many post-translational modification states; however, many challenges still remain, such as optimized antibody scaffolds. Recently, a single-chain fragment antigen binding (Fab) (scFab) format, in which the carboxy-terminus of the light chain is linked to the amino-terminus of the heavy chain, was described to potentially combine the high display levels of a single-chain fragment variable with the high stability of purified Fabs. However, this format required removal of the interchain disulfide bond to achieve modest display levels and subsequent bacterial expression resulted in high levels of aggregated scFab, hindering further use of scFabs. Here, we developed an improved scFab format that retains the interchain disulfide bond by increasing the linker length between the light and heavy chains to improve display and bacterial expression levels to 1-3 mg/L. Furthermore, rerouting of the scFab to the co-translational signal recognition particle pathway combined with reengineering of the signal peptide sequence results in display levels 24-fold above the original scFab format and 3-fold above parent Fab levels. This optimized scFab scaffold can be easily reformatted in a single step for expression in a bacterial or mammalian host to produce stable (Tm of 81 °C), predominantly monomeric (>90%) antibodies at a high yield. Ultimately, this new scFab format will advance high-throughput antibody generation platforms to discover the next generation of research and therapeutic antibodies.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Dominik, Pawel K; Kossiakoff, Anthony A
Phage display selections for affinity reagents to membrane proteins in nanodiscs Journal Article
In: Methods Enzymol, vol. 557, pp. 219–245, 2015, ISSN: 1557-7988.
@article{pmid25950967,
title = {Phage display selections for affinity reagents to membrane proteins in nanodiscs},
author = {Pawel K Dominik and Anthony A Kossiakoff},
doi = {10.1016/bs.mie.2014.12.032},
issn = {1557-7988},
year = {2015},
date = {2015-01-01},
urldate = {2015-01-01},
journal = {Methods Enzymol},
volume = {557},
pages = {219--245},
abstract = {Phage display selections generate high-affinity synthetic reagents that can be used as tools in structural characterization of membrane proteins. Currently, most selection protocols are performed with membrane protein targets in detergents. However, there are numerous technical issues associated with this, primarily that detergents are poor mimics of the native lipid environment. Here, we describe a set of protocols for phage display selection that involves reconstituting membrane proteins in nanodiscs, which are small discoidal particles consisting of lipids enclosed by membrane scaffold proteins. The nanodisc format enabled us to expand the capabilities of competitive and subtractive phage display selection steps, and generation of high-quality synthetic reagents for membrane proteins in native-like lipid environment.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
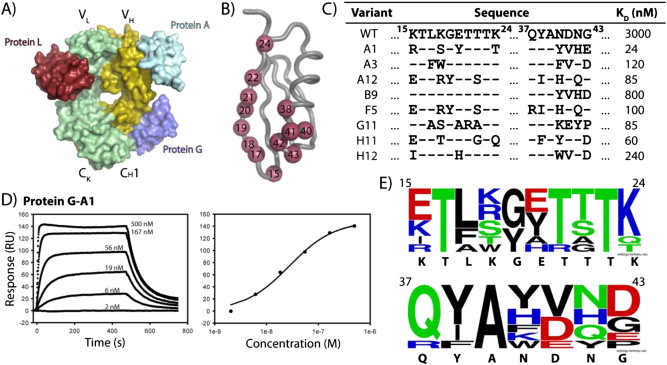
Bailey, Lucas J; Sheehy, Kimberly M; Hoey, Robert J; Schaefer, Zachary P; Ura, Marcin; Kossiakoff, Anthony A
Applications for an engineered Protein-G variant with a pH controllable affinity to antibody fragments Journal Article
In: J Immunol Methods, vol. 415, pp. 24–30, 2014, ISSN: 1872-7905.
@article{pmid25450256,
title = {Applications for an engineered Protein-G variant with a pH controllable affinity to antibody fragments},
author = {Lucas J Bailey and Kimberly M Sheehy and Robert J Hoey and Zachary P Schaefer and Marcin Ura and Anthony A Kossiakoff},
doi = {10.1016/j.jim.2014.10.003},
issn = {1872-7905},
year = {2014},
date = {2014-12-01},
urldate = {2014-12-01},
journal = {J Immunol Methods},
volume = {415},
pages = {24--30},
abstract = {Immunoglobulin binding proteins (IBPs) are broadly used as reagents for the purification and detection of antibodies. Among the IBPs, the most widely used are Protein-A and Protein-G. The C2 domain of Protein-G from Streptococcus is a multi-specific protein domain; it possesses a high affinity (K(D) ~10 nM) for the Fc region of the IgG, but a much lower affinity (KD~low μM) for the constant domain of the antibody fragment (Fab), which limits some of its applications. Here, we describe the engineering of the Protein-G interface using phage display to create Protein-G-A1, a variant with 8 point mutations and an approximately 100-fold improved affinity over the parent domain for the 4D5 Fab scaffold. Protein-G-A1 is capable of robust binding to Fab fragments for numerous applications. Furthermore, we isolated a variant with pH-dependent affinity, demonstrating a 1,000-fold change in affinity from pH7 to 4. Additional rational mutagenesis endowed Protein-G with significantly enhanced stability in basic conditions relative to the parent domain while maintaining high affinity to the Fab. This property is particularly useful to regenerate Protein-G affinity columns. Lastly, the affinity-matured Protein-G-A1 variant was tethered together to produce dimers capable of providing multivalent affinity enhancement to a low affinity antibody fragment-antigen interaction. Engineered Protein-G variants should find widespread application in the use of Fab-based affinity reagents.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}